视神经脊髓炎谱系疾病的临床及影像学特征
2019-03-13王盼盼宋亚雪孟彦宏赵中旻王建华
王盼盼,宋亚雪,孟彦宏,赵中旻,王建华
视神经脊髓炎谱系疾病(neuromyelitis optica spectrum disorder,NMOSD)是一组自身免疫介导的中枢神经系统炎性脱髓鞘疾病,临床和影像学表现多种多样。传统概念认为视神经脊髓炎(neuromyelitis optica,NMO)的典型表现是单侧或双侧视神经炎合并长节段性脊髓炎,无其他部位受累证据的一组临床综合征,即视神经炎与脊髓炎的简单组合。Wingerchuk等[1]研究发现NMO患者血清中存在水通道蛋白4抗体(AQP4-IgG),并且证实部分NMO患者存在颅内非特异性病灶,提出了NMOSD的概念。NMO/NMOSD的临床表现比既往所认识到的更为丰富,其影像学也存在较大差异,且伴发疾病各不相同。本文对我院收治的9例NMOSD病例进行报道,以加深对此综合征的认识。
1 临床资料
1.1 一般资料 9例患者均是2013年6月-2017年6月河北省人民医院神经内科收治的符合2015版诊断标准的NMOSD患者[2]。其中AQP4-IgG阳性5例,AQP4-IgG阴性或未行抗体检测的4例。男3例,女6例,男女比例3∶6。就诊年龄:20~77岁,平均48.8岁。9例患者主要临床表现为视力下降、顽固性呃逆、恶心、呕吐(极后区综合征),肢体无力、浅感觉减退或消失、神经性疼痛、痛性肌痉挛、二便潴留、体位性低血压、以及复视、眩晕、构音障碍、共济失调(急性脑干综合征)等,症状可先后发生或者叠加存在,临床表现个数1~6个,平均3个。首次发病表现为视力减退,诊断为视神经炎2例(2/9),其中1例5 y后出现脊髓受累(AQP4-IgG阳性),另1例在20 y后出现脊髓受累(AQP4-IgG未查),符合传统的NMO和新标准NMOSD,此2例患者至最后一次随访时颅内未见受累。另外7例患者至最后一次随访时5例均以肢体麻木、感觉异常、疼痛或二便异常为主要表现;其余2例表现为眩晕、复视伴肢体麻木(其中1例复发时出现了视力障碍)。此7例患者中,3例脊髓和颅内组织均受累(AQP4-IgG均阳性),4例只有脊髓受累(1例AQP4-IgG阳性,2例AQP4-IgG阴性,1例未行抗体检测)。其中1例AQP4-IgG阳性患者首次发病表现为左侧脊髓半切综合征,6个月后复发时表现为右侧脊髓半切综合征,此例只有脊髓受累,无视力下降和颅内组织受累(见表1)。
1.2 MRI检查及实验室结果(见表2) 9例患者中,病灶仅在颈段脊髓4例,胸段脊髓3例,病变同时累及颈段、胸段脊髓2例。其中2例表现为极后区综合征的患者病灶累及延髓和颈髓交界区。髓内病灶表现为长T1、长T2信号,FLAIR序列高信号。7例患者增强后呈亮斑样、斑片样或线样强化。例2患者胸椎MRI表现为完全横贯性损害,伴轻中度强化(见图1),例4患者两次发作表现为典型脊髓半切,第一次发作时呈颈髓左侧半切;第二次发作呈右侧颈髓半切(见图2),均伴有不规则强化。3例患者有颅内异常信号:(1)中脑导水管及四脑室、侧脑室和脑干周围软脑膜受累2例,其中1例为侧脑室室管膜“铅笔杆”样强化,同时伴脑干周围软脑膜“线性”强化(见图3);另1例表现为MRI平扫中脑导水管、四脑室周围异常信号,无明显强化(见图4)。(2)侧脑室旁及大脑半球白质异常信号1例,表现为皮质下白质散在点状或片状融合病灶,无明显强化(见图5)。此3例患者均伴有脊髓受累。7例患者进行了AQP4-IgG检测(采用国际上推荐的间接免疫荧光法IIFT),阳性5例,阴性2例,2例未行AQP4-IgG抗体检测;7例患者寡克隆带(OB)均为阴性。9例患者脑脊液均为清亮透明,压力正常8例,1例升高(270 mmH2O);白细胞正常7例,升高2例(分别是79×106、22×106);蛋白正常7例,升高2例(分别是100.44 g/L、51.87 g/L);糖正常7例,升高2例(分别为86.91mmol/L、143.97 mmol/L),氯正常9例。9例患者中,1例表现为抗中性粒细胞胞浆抗体、抗中性粒细胞胞浆抗体(核周型)弱阳性,抗核抗体阳性;另1例仅抗核抗体谱阳性。促甲状腺激素(TSH)升高者1例(6.76 mmol/L),9例甲状腺相关抗体(甲状腺球蛋白抗体、甲状腺过氧化物酶抗体)均正常;甲状腺功能及血清肿瘤标记物未见异常。
1.3 治疗及预后 9例患者均使用糖皮质激素冲击治疗(1000 mg×3 d,500 mg×3 d,240 mg×3 d,120 mg×3 d,60 mg×7 d,50 mg×7 d,顺序递减直至中等剂量30~40 mg/d),4例患者在急性期应用大剂量丙种球蛋白治疗,5例患者在激素冲击治疗后加用硫唑嘌呤2 mg/kg/d。
所有患者均进行随访,时间9~39个月,平均12.1个月。随访结果发现,9例患者均遗留有不同程度后遗症,视力下降4例,肢体麻木、无力5例。截至到最后一次随访,复发患者5例,未复发者4例。复发时再次激素冲击治疗5例,丙种球蛋白治疗2例,治疗后症状均有不同程度好转。

表1 9例NMOSD患者一般资料
注:ATM:Acute transverse myelitis,急性横贯性脊髓炎;ON:Optica neuritis,视神经炎

表2 9例NMOSD患者影像学及AQP4-IgG结果
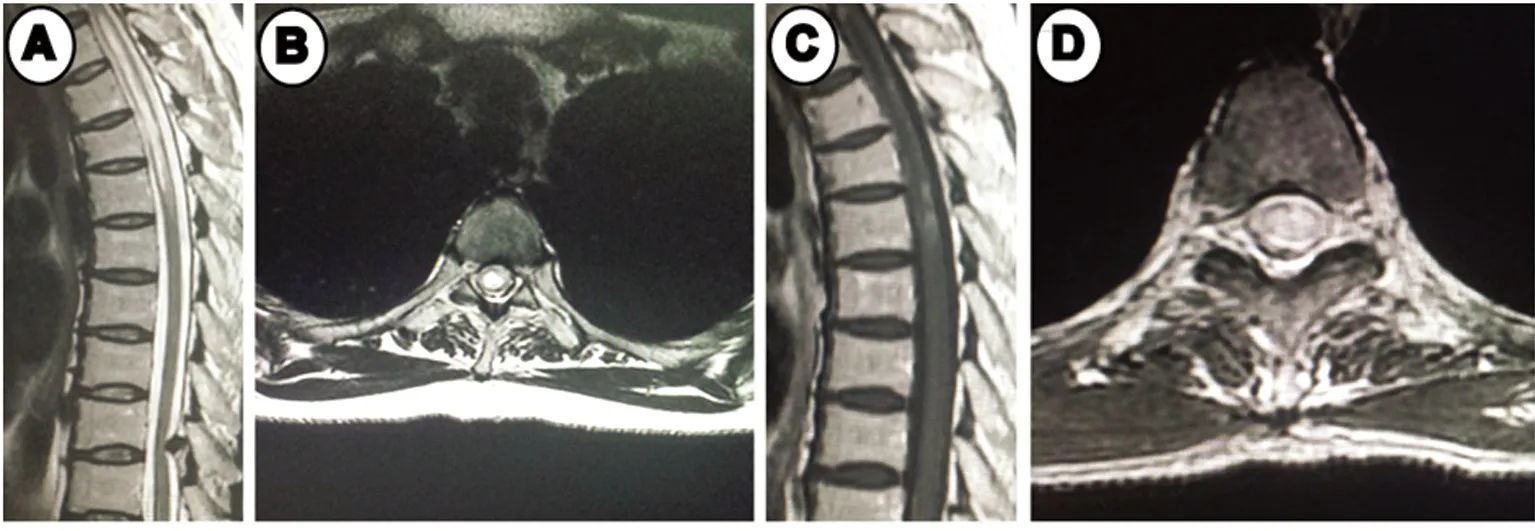
图1 胸椎MRI。A、B:T2WI示T2-5高信号;C、D:T2-5髓内异常信号影,周边可见轻中度强化,边界不清
图2 第一次发病:颈椎MRI。A、B:T2WI示C2-5椎体水平脊髓左侧高信号影;C、D:C2-5脊髓左侧小片状轻中度强化,边界不清。 第2次发病:颈椎MRI。E、F:T2WI示C2-5椎体水平脊髓右侧异常信号影;G、H:C2-5椎体水平右侧小片状高信号强化
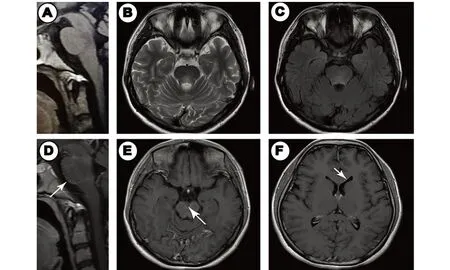
图3 头部+颈椎MRI。A:T1WI未见异常信号;B、C:T2WI及Flair相示脑桥背侧高信号;D、E:脑桥周围软脑膜线性强化(白色箭头所示);F:左侧侧脑室“铅笔杆样”强化(白色箭头所示)
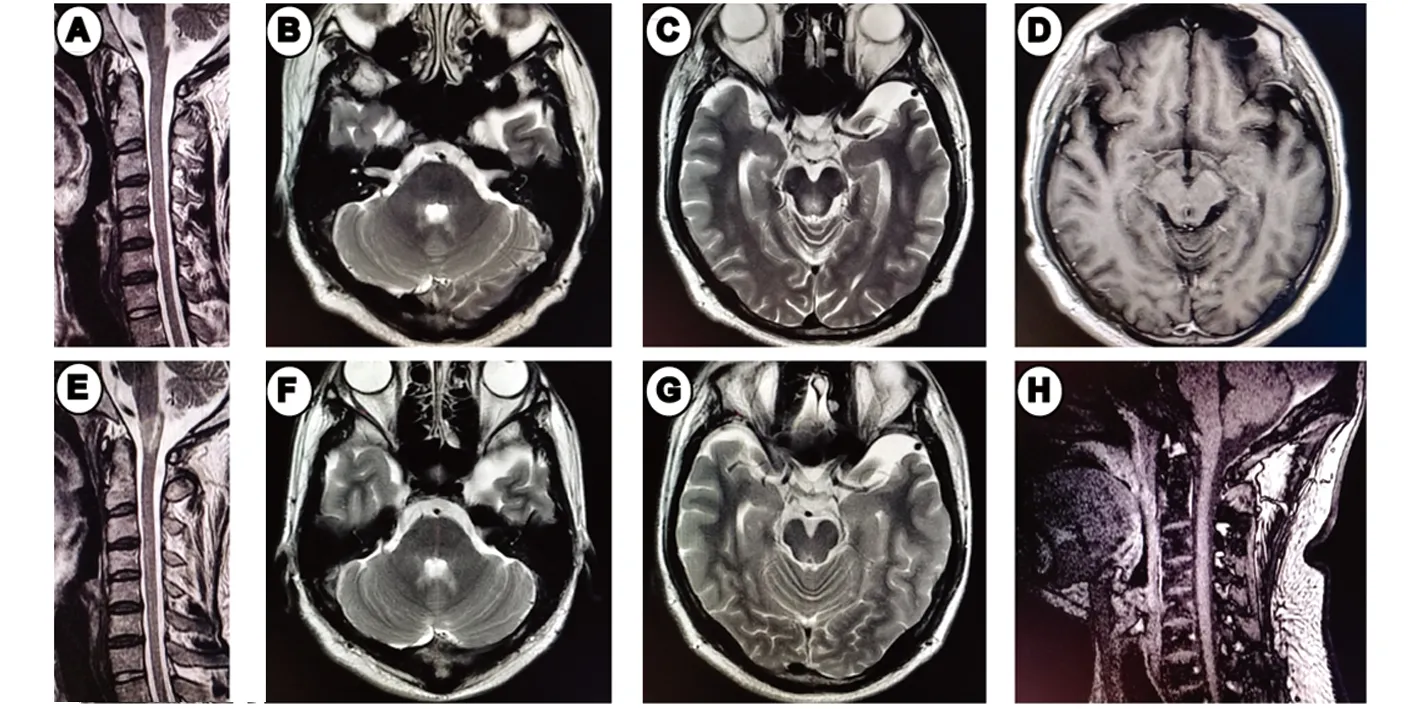
图4 第一次发病:颈椎+头部MRI。A:T2WI无异常信号影;B、C:T2WI四脑室、中脑导水管周围高信号;D:病灶无强化。第2次发病:颈椎+头部MRI。E:T2WI示延髓与上颈段交界区高信号;F、G:四脑室、中脑导水管病灶范围较前缩小;H:延髓与上颈段交界区病灶未见强化

图5 颈椎+胸椎+头部MRI。A、B:T2WI示高颈段脊髓高信号;D、E:T2WI示T7-8水平脊髓内片状高信号;C、F:Flair示大脑半球及侧脑室异常信号
2 讨 论
NMO多以严重的视神经炎与纵向横贯性脊髓炎为特征性表现。近10余年的研究发现NMO的临床表现更为广泛,包括颅神经受累、意识改变和一些异质性较大的非特异性症状。NMO-IgG的发现使我们逐渐认识到NMO是独立于多发性硬化(Multiple sclerosis,MS)的疾病实体,从而提出了NMOSD的概念。
NMOSD临床表现多种多样,除最具特征性的ON、急性脊髓炎和极后区综合征以外,还有其他脑病类型。(1)ON多累及视神经后段及视交叉,病变长度多大于1/2视神经长度。本研究中2例以视力下降发病,由于就诊时视神经处于非活动期,影像学上未见明显异常。(2)脊髓炎多累及中央灰质和部分白质,矢状位上多呈连续病变,其纵向延伸长度往往超过3个椎体节段,MRI轴位上常表现为点片状、“H”形、半横贯或完全横贯性病灶。本研究中1例患者表现为两次典型脊髓半切综合征,MRI分别显示为左、右典型半横贯性损害,极为少见。(3)极后区的血-脑屏障相对薄弱,更容易受AQP4-IgG攻击[3],受损后出现顽固性呃逆、恶心、呕吐;MRI表现为延髓背侧受累,主要累及极后区,呈片状或线状长T2信号。Misu等[4]首次提出了“线样延髓”损害的概念,即延髓病变与颈髓病变相连,随后更多的研究发现延髓、颈髓连续病灶是NMOSD较为特异的表现[5~7]。本研究中有2例患者的临床表现符合极后区综合征,影像学改变为典型延髓、颈髓连续性病损,支持以上文献报道。(4)急性脑干综合征主要表现为头晕、复视、共济失调等,部分病变无明显临床表现。Li等[8]报道了1例39岁亚裔以多种脑干症状发病的女性患者,表现为顽固性呃逆、呕吐、眩晕、复视、眼球运动障碍及味觉减退等多颅神经受累。本研究中有2例患者以脑干综合征发病,表现为常见脑干受损的临床症状,其中1例还出现了难治性瘙痒[9]、味觉减退[9]、突发听力下降等[10]罕见的脑干综合征表现。孤束核和中央被盖束受累可能会引起味觉障碍,脊髓背侧、三叉神经脊束及中脑导水管周围受累可能与瘙痒有关[9,10]。(5)急性间脑综合征主要表现为嗜睡、发作性睡病、低钠血症及体温调节异常等,部分病变无明显临床表现。(6)急性大脑综合征主要表现为淡漠、反映迟钝、认知水平下降和头痛等,部分病变无明显临床表现。本研究中有1例患者头部MRI显示大脑半球异常病灶,但没有相应的临床表现,推测可能是非活动期病损或非特异性病灶。
仅表现为脑干综合征的NMOSD患者发病时更趋向年轻[11],AQP4-IgG介导的脑病/脑干综合征发病年龄也较早,表明NMOSD可能具有年龄依赖的解剖结构易感性或某一器官与AQP4抗体结合程度不同的特性。Long等[12]研究发现,NMOSD首次发病无ON和脊髓炎表现的患者发病年龄更年轻,复发出现较早,有更多的颅内受累症状。本研究20岁女性(例5)和25岁男性(例7)患者在首次发病时均表现为急性脑干综合征;前者首次发病1个月后出现四肢麻木、颈后部瘙痒和双手活动笨拙等,3个月后出现右眼视物模糊,1 y后出现眩晕、复视、构音障碍、吞咽困难等,该患者先后出现脑干综合征、脊髓炎、ON和脑干综合征;后者首次发病后6个月出现顽固性呃逆、呕吐,伴有背部及双手麻木,MRI显示延髓及高颈髓异常信号,符合极后区综合征。此2例患者的发病及病情发展支持年轻患者易复发及更多颅内受累的观点。
总之,NMOSD首次发病见于各年龄段,对急性或亚急性出现的视物不清、恶心呕吐、呃逆、运动感觉障碍、合并自主神经功能异常及意识改变等,需考虑有无NMOSD可能,应及时检测血清及脑脊液AQP4-IgG,根据最新诊断标准作出诊断。临床医生应加深对NMOSD临床及影像学特征的认识,以利于早期诊断和早期治疗,从而达到减少复发、改善预后和提高患者生活质量的目的。
