马来酸酐接枝PS对PS/PLA共混体系发泡行为的影响
2019-02-26王亚桥江翰川霓敬越陈剑泽王向东
王亚桥,江翰川,霓敬越,陈剑泽,王向东
(北京工商大学材料与机械工程学院,北京 100048)
0 前言
挤塑聚苯乙烯泡沫塑料(XPS)的绝热性能优异、吸水率较低、尺寸稳定性较好、在墙体保温、土木工程、冷链物流等领域有广泛应用[1]。氢氯氟烃(HCFCs)被广泛用作聚苯乙烯挤出发泡板材的发泡剂,但是HCFCs属于臭氧消耗物质(ODS),同时也是温室气体,因而HCFCs即将被淘汰。对环境友好和气候友好的CO2组合发泡技术被选为最主要的替代技术[2-3]。然而,以CO2为发泡剂时,由于其与聚苯乙烯的相容性差,致使其在聚苯乙烯中溶解度低,扩散速率快,难获得低密度制品[4-6]。将和CO2极性相近的聚合物与PS共混是一种提升CO2的溶解度有效方法[7-9]。同时作为分散相的聚合物与PS的界面也可以作为气泡成核点,促进气泡成核[10-11]。PLA是一种对CO2吸附能力较强的聚合物,本文采用熔融共混法制备PS/PLA共混物,以提高CO2在共混体系中的溶解度,进而研究了PS-g-MAH对PS/PLA共混体系的CO2溶解度、流变行为、相态结构以及发泡行为的影响。
1 实验部分
1.1 主要原料
PS,158K,BASF-YPC有限责任公司;
PLA,2003D,NatureWorks股份有限公司;
PS-g-MAH,PS-35,BASF有限责任公司;
CO2,纯度>99.5 %,北京氧立来气体有限公司。
1.2 主要设备及仪器
转矩流变仪,XSS-300,上海科创橡塑机械设备有限公司;
压片机,LP-S-50,瑞典Labtech Engineering公司;
旋转流变仪,MARS,美国TA仪器公司;
高压发泡釜装置,200 ml,北京森郎科技有限公司;
密度天平,CPA2245,赛多利斯科学仪器有限公司;
扫描电子显微镜,FEG250,美国FEI公司。
1.3 样品制备
按照表1的配方在温度为190 ℃,转速为50 r/min的条件下,在密炼机中进行熔融共混10 min,压片制样;将超临界CO2注入发泡釜中,并分别在134 ℃,12.5 MPa下稳定3 h,使CO2充分溶入PS树脂中,然后打开发泡釜的泄气阀将压力瞬间释放,得到所需的PS泡沫。
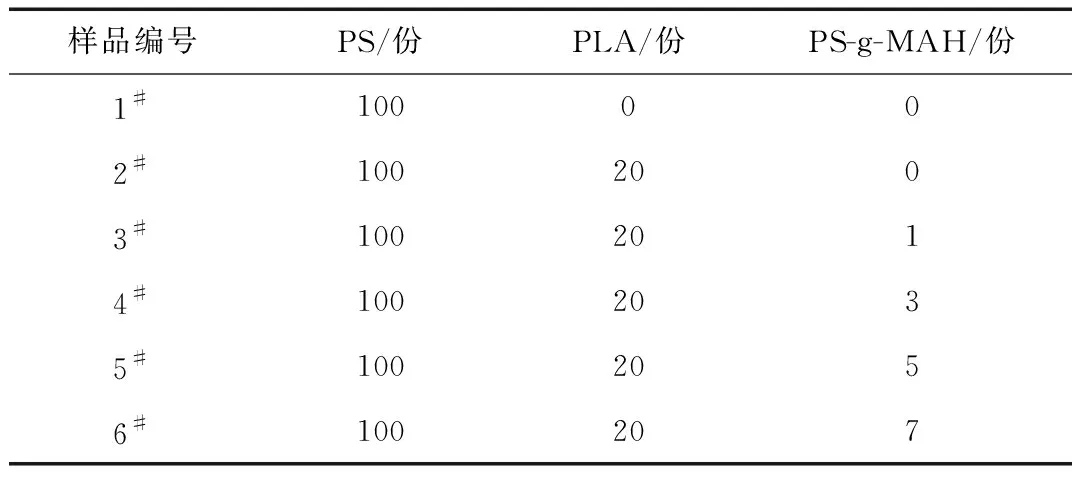
表1 PS/PLA/PS-g-MAH共混物的配比
1.4 性能测试与结构表征
旋转流变仪测试:采用旋转流变仪对各个样品的动态剪切流变性能进行研究,取得直径15 mm,厚度为2.0 mm的实验试样放于平行板夹具间,测试温度为190 ℃;剪切频率范围为0.1~100 Hz,最大应变应力为5 %;
溶解度测试:将相同质量为0.3 g的样品在50 ℃、12.5 MPa的条件下在高压釜中浸泡10 h,同样以100 ℃、12.5 MPa条件在高压发泡釜浸泡5 h后,打开发泡釜并的瞬间开始计时并测量记录样品质量变化,并以Origin软件统计计算获得溶解度[12];
密度测试:采用密度天平对各个发泡样品的密度(ρfoam)进行表征,每组样品测试3次,通过式(1)计算;
(1)
式中Wa——天平上测得的样品质量,g
Wfl——样品排水质量和样品质量的差值,g
ρfl——测试时刻水的密度,g/cm3
SEM测试:在液氮中脆断后,对泡沫断面喷金,设置电压10 kV,观察发泡样品微观形貌;
泡孔统计:泡孔尺寸和泡孔密度统计:在Image-pro Plus中对泡孔的尺寸进行统计计算得到平均尺寸,并得到泡孔单位面积上的泡孔数,泡孔密度由式(2)~(3)计算得到[13]。
(2)
(3)
式中φ——发泡倍率
ρp——发泡前样品密度,g/cm3
ρf——发泡后样品的密度,g/cm3
n——泡孔密度,个/cm3
nb——统计面积中的泡孔数量,个
L——统计面积中的边长,cm
2 结果与讨论
2.1 PS/PLA共混体系CO2的溶解度
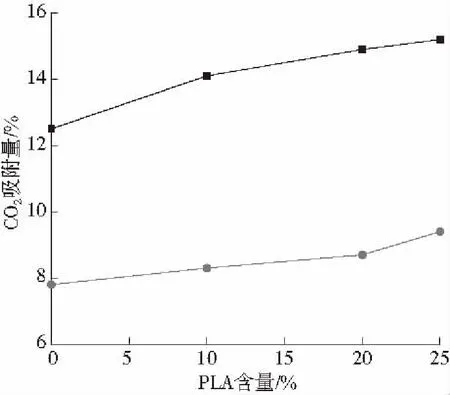
浸泡温度/℃:■—50 ●—100图1 不同条件下CO2在PS/PLA体系中的溶解度Fig.1 The solubility of CO2 in PS/PLA blends at different temperature
从图1中可以看出随着PLA含量的增加,共混体系溶解度逐渐提高。这是因为PLA中含有羰基基团,在吸附CO2的过程中羰基作为电子的供体,CO2作为电子的受体,因此两者具有较强相互吸引作用,导致共混体系的CO2溶解度升高。当PLA含量为25份时,CO2的溶解度比纯PS提高了22 %。高温时CO2在共混体系中的溶解度要低于低温时的溶解度,这是因为CO2在高温时的扩散速率增加,需要更大的压力才能使CO2在PS中的溶解于扩散达到平衡,因此在相同的压力下,高温时CO2在聚合物中的溶解度低。
表2是PLA为20份时不同含量PS-g-MAH时CO2在PS/PLA体系中的溶解度。可以看出随PS-g-MAH含量的增加,CO2在PS/PLA体系中的溶解度变化不大。

表2 不同含量PS-g-MAH时CO2在PS/PLA体系中的溶解度
2.2 PS/PLA/PS-g-MAH共混体系的反应情况
从图2中可看出随PS-g-MAH接枝量的增加,扭矩增加。这是共混体系的黏度增加造成的,PS-g-MAH中的马来酸酐基团在一定的条件下容易被开环并转换为羧酸基团, 并与PLA上的羟基的进行反应,形成PS-g-PLA,这种结构使体系黏度提高。同时,PS-g-PLA结构还降低了两相界面的界面张力,减小分散相的粒径。
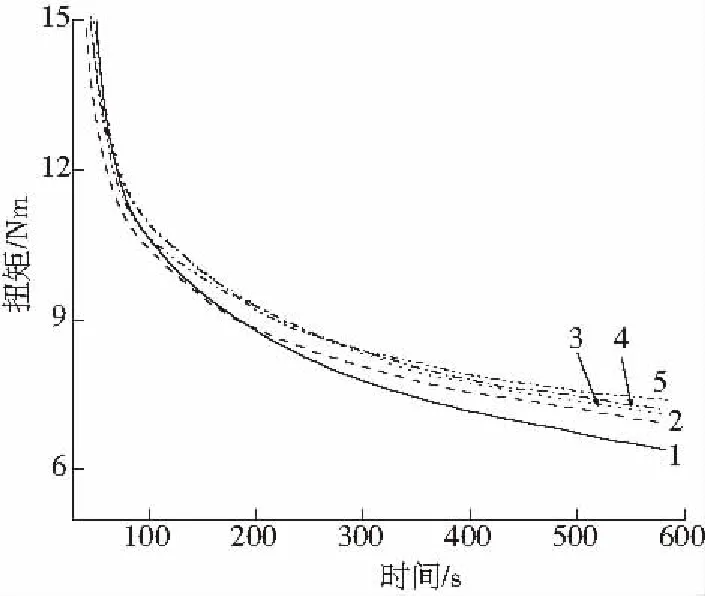
1—2# 2—3# 3—4# 4—5# 5—6#图2 随PS-g-MAH含量的增加共混物扭矩的变化Fig.2 Torque of PS/PLA blends with different PS-g-MAH contents
图3是PS-g-MAH以及PS-g-MAH增容的共混体系的FTIR谱图。在图中1 854 cm-1和1 780 cm-1处的峰分别代表马来酸酐上羰基的对称振动及反对称振动;PS-g-MAH与PS/PLA熔融共混后,酸酐环被打开形成2个羧酸基团。而1 854 cm-1和1 780 cm-1处的峰消失,证明马来酸酐与PLA的端羟基发生了反应。
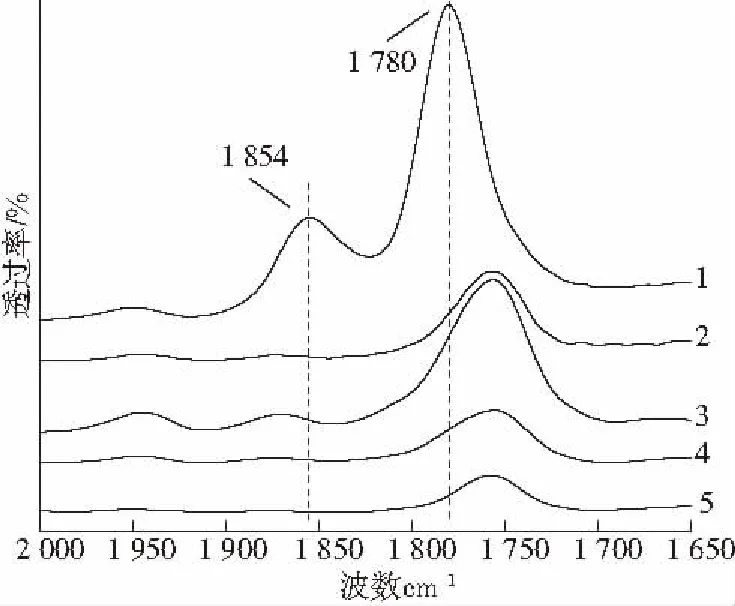
1—2# 2—3# 3—4# 4—5# 5—6#图3 PS-g-MAH和共混体物FTIR谱图Fig.3 FTIR spectrum of PS-g-MAH and PS/PLA/PS-g-MAH blends
图4是随着PS-g-MAH含量的增加,PS/PLA共混体系相态结构的变化。随着PS-g-MAH含量的增加,PLA分散相平均尺寸明显减小,如图5所示,分散相平均尺寸从1.178 μm减小到0.451 μm。证明PS-g-MAH与PLA反应形成了PS-g-PLA,增加了PS/PLA的相容性。但PS-g-MAH含量超过3份后,分散相的平均粒径下降幅度开始减小。随着PS-g-MAH含量的增加而增加,分散相的断面密度显著增加。当PS-g-MAH含量从0份增加到7份时,断面密度从1.45×105个/cm2增至11.9×105个/cm2。分散相尺寸的减小,数量的增加,有利于发泡过程中泡孔成核。

PS-g-MAH含量/份:(a)0 (b)1 (c)3 (d)5 (e)7图4 随PS-g-MAH含量的增加共混物的相态变化(×10 000)Fig.4 Dispersion phase change with different PS-g-MAH contents
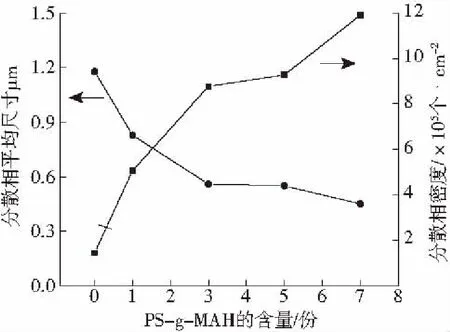
图5 分散相平均尺寸和密度随PS-g-MAH含量的变化Fig.5 Variety of dispersion phase with different content of PS-g-MAH
2.3 PS/PLA共混体系的流变性能
聚合物的储能模量是衡量聚合物熔体弹性的关键指标。图6(a)为PS/PLA共混体系的储能模量和频率的曲线。聚合物的可发性与储能模量有密切地关,一般认为储能模量越大,聚合物的熔体弹性就越好, 熔体的可发性也越好。图中的PS/PLA共混体系的储能模量低于纯PS。这主要是因为PLA树脂本身的熔体弹性低于PS,以及PS与PLA的相容性差,界面结合力弱,在剪切易发生脱落,所以引入PLA后共混体系的储能模量下降。当PS/PLA共混体系中加入PS-g-MAH后,PS/PLA共混体系的储能模量随着PS-g-MAH含量的增加升高。这是因为PS-g-MAH中的酸酐基团可与PLA中的端羟基反应,生成PS-g-PLA,改善了PS/PLA两相的相容性。使两相界面结合力加强因此PS/PLA共混体系的熔体粘弹性增加。图6(b)为PS/PLA共混体系的复数黏度与剪切频率的曲线。从图6(b)中可已看出,各个样品的复数黏度表现出典型的剪切变稀现象。在低频区,PS的复数黏度高于PLA,但随着PS-g-MAH含量的增加,PS/PLA复数黏度逐渐增加。这是因为PS-g-MAH中的酸酐基团可与PLA中的端羟基反应,生成PS-g-PLA,改善了PS/PLA两相的相容性,使两相的界面黏着力增强。
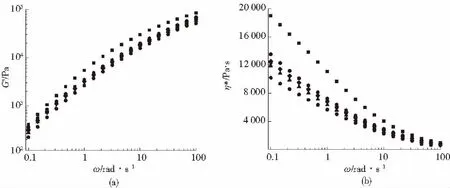
■—1# ●—2# ▲—3# ▼—4# ◆—5# —6#(a)G′-ω曲线 (b)η*-ω曲线图6 PS/PLA/PS-g-MAH共混体系动态流变性能Fig.6 Rheological property of PS/PLA/PS-g-MAH blends
2.4 PS/PLA共混体系的发泡行为
图7为PS/PLA共混体系的泡孔形态,表3为泡沫样品结构参数。纯PS泡沫的泡孔尺寸为142.01 μm,泡孔壁厚。当加入20份PLA后,泡孔泡孔尺寸明显下降,泡孔密度从7.62×107个/cm3增至1.52×108个/cm3,上升了一个数量级,泡沫密度由0.087 g/cm3下降到0.076 g/cm3。这是因为PS-g-MAH 有效改善了PS与PLA的相容性,使分散相的粒径减小,两相界面数量增加,而两相界面处可作为气泡成核点,利于气泡成核,实验结果表明两相界面起到了气泡成核点的作用。

(a)1# (b)2# (c)3# (d)4# (e)5# (f)6#图7 PS/PLA/PS-g-MAH共混体系泡沫的泡孔形态(×400)Fig.7 SEM micrographs of PS/PLA/PS-g-MAH foams(×400)
从图8中可以观察到泡孔壁上大量的分散相。这是因为泡孔成核后,溶解在PLA中的CO2扩散到泡孔中,PLA分散相并未发泡,并在泡孔增长完成后,PLA分散相分布并固化在泡孔壁上。

图8 PS-g-MAH为7份时泡孔壁上的PLA分散相(×2 000)Fig.8 PLA phase in cell wall with 7phr PS-g-MAH
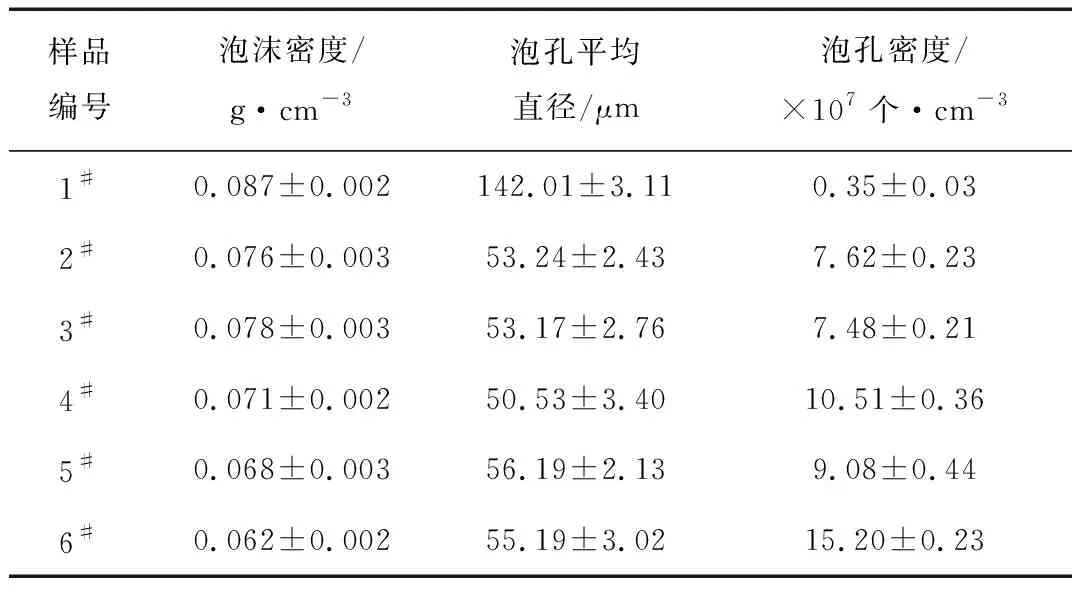
样品编号泡沫密度/g·cm-3泡孔平均直径/μm泡孔密度/×107个·cm-31#0.087±0.002142.01±3.110.35±0.032#0.076±0.00353.24±2.437.62±0.233#0.078±0.00353.17±2.767.48±0.214#0.071±0.00250.53±3.4010.51±0.365#0.068±0.00356.19±2.139.08±0.446#0.062±0.00255.19±3.0215.20±0.23
3 结论
(1)随着PLA含量的增加,PS/PLA共混体系的CO2溶解度提升,当PLA含量达到25份时,共混体系的CO2溶解度提高了22 %;
(2)随着PS-g-MAH 含量的增加PLA分散相尺寸明显下降,从1.178 μm下降到0.451 μm,且分散相数量增多;
(3)随着PS-g-MAH 含量的增加,PS/PLA共混体系的储能模量和复数黏度逐渐增加;PS/PLA共混体系的泡沫的泡孔密度从7.62×107个/cm3增至1.52×108个/cm3,表明两相界面起到了气泡成核的作用,同时泡沫密度降低。
