天麻中天麻素标准物质的研制
2019-02-23初侨席兴军董跟来赵新颖王尉王风贺张维冰兰韬
初侨,席兴军,董跟来,赵新颖,王尉,王风贺,张维冰,兰韬*
(1.中国标准化研究院农业与食品标准化研究所,北京 100191;2. 亚东信基(北京)农产品有限公司,北京 100125;3.北京市理化分析测试中心,北京 100089;4. 南京师范大学环境学院,江苏 南京 210023;5. 华东理工大学化学与分子工程学院,上海 200237)
天麻为我国常用名贵中药材,是多年生兰科草本植物天麻的干燥根块,主产于贵州、陕西、云南、四川、湖北等地,具有平肝息风、通络止痛的功效,主治风痫、惊气等引起的头风头痛、半身不遂、小儿惊痫动风、风湿痹症[1-2]。天麻中含有天麻素、天麻苷元、天麻醚苷等成分[3-4],其中天麻素是天麻的主要有效成分,含量通常可高达0.33%~0.67%,具有较好的镇静、安神作用,常被用作天麻药材质量控制的指标性成分[5-9],其化学结构为4-羟甲基苯甲醇-β-D-吡喃葡萄糖苷,结构式如图1所示[10-12]。
标准物质具有可在时空两个领域进行量值传递的特征,因此在分析方法评价、测量结果及仪器验证的准确性保证方面具有重要作用,对于分析化学和临床分析尤其重要[13-16]。天然产物类标准物质可以为我国特有的中药材产品质量控制和检测结果的准确性、可比性和溯源性提供保证[17-18]。此类标准物质主要通过分离精提的方法从中药材或其提取物中制备,使目标产物的纯度达到98%以上,经定性确定为该目标物后再经均匀性、稳定性检验和定值分析制备获得。随着中医药以及保健品行业的复苏,社会对此类标准物质的需求也在不断增加。而天然产物类标准物质大多来源于中草药提取物,存在不同来源、种类、结构的中草药有效成分标准物质制备方法差异较大、提取工艺复杂、成本高、某些成分稳定性较差等问题,制约了此类标准物质的发展。
以天麻素为代表的标准物质价格较为昂贵,为了扩充标准物质的种类,降低标准物质的使用成本,满足天麻等相关产品质量控制工作的需求,本文依据标准物质研制技术规范(JJF 1343—2012)[19]以及 ISO导则34、35的技术要求[20-21],制备了天麻素标准物质(图1),以紫外、红外、质谱、核磁共振波谱等4种方法对制备的天麻素标准物质进行定性分析,同时以高效液相色谱方法进行均匀性和稳定性检验,最后对该标准物质进行了定值分析。
1 仪器与材料
1.1 主要仪器与试剂
EClassical 3100高效液相色谱仪(大连依利特分析仪器有限公司);LC-6AD型制备液相色谱仪(日本岛津公司);HT7100系列制备液相色谱系统(苏州汇通色谱分离纯化有限公司);Spectrum 400傅里叶变换红外光谱仪(美国珀金埃尔默公司);UV-4501型紫外可见分光光度计(天津港东科技发展股份有限公司);1100 Series LC/MSD Trap高效液相色谱-质谱仪(美国安捷伦公司);Bruker 400 MHz核磁共振波谱仪(德国布鲁克公司);Milli-Q超纯实验室用水机(德国默克密理博公司)。
天麻:市购;甲醇、乙腈:色谱级,德国默克公司;实验所用其他试剂均为分析纯;超纯水为实验室自制。
1.2 样品制备
首先以兰科植物天麻GastrodiaelataBl.的干燥块茎为原料,将其粉碎至100目粉末,精密称取10 g置索氏提取器中,加无水乙醇60 mL,连续回流6 h,回收无水乙醇,残渣用无水乙醇溶解滤过,滤液置于50 mL容量瓶中,用少量无水乙醇多次洗涤容器和滤器,合并洗涤液,用无水乙醇定容,摇匀。向其中加入30 mL石油醚,搅拌30 min,搅拌结束后静置10 min,分液得到下层脱脂后的溶液,将脱脂后的溶液旋转蒸干得浸膏,加入石油醚定容到10 mL,加入5 mL水饱和的正丁醇,搅拌10 min,搅拌结束后静置分液,取下层正丁醇层得天麻提取液。

1.3 定性方法
制备的天麻素高纯单体化合物经紫外光谱分光光度法、傅里叶变换红外-近红外光谱法、高效液相色谱-质谱法和核磁共振波谱法定性。紫外-可见分光光度计测定提取物的紫外光谱时以乙腈为溶剂,于0~400 nm波长下扫描得提取物的紫外光谱图。采用KBr压片法制备提取物的红外检测样品,以傅里叶变换红外-近红外光谱仪测定提取物的红外光谱图。采用高分辨飞行时间质谱仪在正、负离子模式下测定提取物的质谱图。以DMSO-d6(含TMS)为氘代试剂,采用400 MHz核磁共振波谱仪测定得到25 ℃下提取物的氢谱和碳谱。
1.4 纯度定值方法

2 方法与结果
2.1 天麻中天麻素的提取
以天麻提取液为原料,采用制备液相色谱制备得到天麻素精提物,以天麻素提取液、天麻素精提物以及天麻素标准品为样品进样,以1.4中纯度定值方法中的色谱条件进行色谱分析,结果如图2所示。由图2可知,天麻素标准品色谱峰的保留时间与天麻提取液中保留时间在5.08~5.43 min内的色谱峰相一致,说明此峰为天麻素的色谱峰,制备得到的天麻素精提物的保留时间也与天麻素对照品的保留时间一致,且纯度较高。

图2 天麻素提取液、天麻素精提物和天麻素标准品色谱图Fig.2 Chromatogram of the extract of gastrodin, refined extract of gastrodin and control gastrodin
2.2 定性分析
制备的天麻素高纯提取物经紫外光谱分光光度计、傅里叶变换红外-近红外光谱仪、高效液相色谱-质谱仪和核磁共振波谱仪完成定性。
天麻素精提物的紫外谱图如图3所示,在220 nm处有最大吸收波长。

图3 天麻素紫外光谱图Fig.3 Ultraviolet spectrogram of gastrodin
天麻素正离子模式下质谱图如图4所示,检测结果:m/z=287.128 5 [M+H]+,m/z=573.128 6[2M+H]+,结果与天麻素的分子量286.227 8相符。

图4 天麻素正离子模式下质谱图Fig.4 Mass spectrogram of gastrodin under positive ion mode
天麻素的1H-NMR(400 MHz,DMSO-d6)δ: 6.97 (2H, d,J= 8.4 Hz, H-2, 6), 7.22 (2H, d,J= 8.4 Hz, H-3, 5), 4.42 (2H, s, H-7), 4.82 (1H, d,J= 7.2 Hz, H-1′), 3.21~3.29 (4H, m, Glc-H-2′, 3′, 4′, 5′), 3.68 (1H, d,J= 11.4 Hz, H-6a′), 3.45 (1H, dd,J= 4.8, 2.0 Hz, H-6b′);13C-NMR(400 MHz,DMSO-d6)δ:156.8 (C-1), 116.4 (C-2, 6), 128.2 (C-3, 5), 136.3 (C-4), 63.0 (C-7), 101.0 (C-1′), 73.7 (C-2′), 77.5 (C-3′), 70.2 (C-4′), 77.1 (C-5′), 61.2 (C-6′),与文献[22]中数据一致。
2.3 均匀性检验
根据JJF 1343—2012《标准物质定值的通用原则及统计学原理》[19]标准中对均匀性检验方法的要求,对制备的天麻素标准物质的均匀性进行检验。采取随机抽样、多次测量的策略,从天麻素标准物质分装的200个小瓶中随机抽取其中11小瓶,每瓶中取样3次,每次取样量为5 mg,用色谱甲醇配制为1 mL溶液,进行高效液相色谱分析,以色谱峰面积所占百分比作为样品的纯度值,进行瓶内和瓶间的均匀性检验,结果如表1、表2所示。

表1 天麻素均匀性检验的测定数据

表2 天麻素标准物质均匀性研究方差分析结果
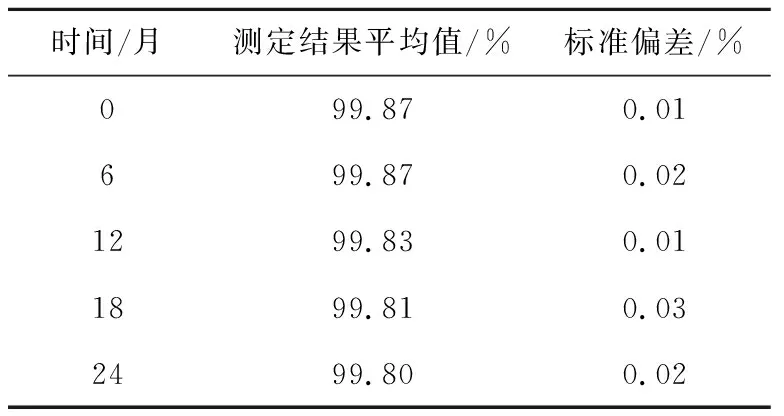
由表2可知,当υ1=10,同时υ2=22时,查F表,得F0.05(10,22)=2.30,由于F= MS间/MS内=1.77 制备的天麻素标准物质在样品分装后,于4 ℃保存。本文考察了制备后的天麻素标准物质在两年内的稳定性,每6个月取样一次进行检测,方法同均匀性检验,每个样品检测5次,以5次检测结果的平均值为检测结果,如表3所示。 表3 长期稳定性检验实验结果(n=5) 从天麻素标准物质的长期稳定性数据可以看出,样品的纯度随着时间推移略有下降,但降低非常不明显,仍处于正常范围内,以直线的线性关系为经验模型,考察线性关系的斜率随标准物质稳定性变化的变化值,以此变化值对标准物质的稳定性进行预测。该线性关系的斜率值计算如下: 直线上的点的标准偏差为: 与斜率相关的不确定度为: 自由度为n-2和p=0.95(95%置信水平)的分布因子t等于3.182。 由于|β1| 该标准物质长期稳定性不确定度的贡献为:us=s(β1)×t=0.05%。 所制备的天麻素标准物质经8家实验室联合实验定值,本次参加定值的实验室均为北京市内高校和科研院所的分析实验室,分别为北京市理化分析测试中心、岛津分析中心、中国科学院过程工程研究所、北京工业技师学院、北京化工大学、首都师范大学、北京工商大学和中国农业科学院饲料研究所。该标准物质的定值方法为随机抽取16瓶样品,每个参与定值的实验室随机送样2瓶,每瓶样品采样3次,进行3次测定,每家参与实验室提供6个测量数据,本次定值分析8家参与实验室均采用高效液相色谱-面积归一化法的定值方法,每家实验室在该方法的基础上选取的色谱条件略有改变,各实验室汇总后的定值数据及其方差分析结果如表4所示。 首先,对8组纯度定值结果进行格拉布斯(Grubbs)检验以剔除可疑值,计算得Gmax=1.212;Gmin=1.515,查格拉布斯临界值表知:G0.05(8)=2.126。因Gmax、Gmin均小于G0.05(8),检验结果为合格。 由于高效液相色谱-面积归一法具有一定的局限性,故分别采用卡尔·费休法、灼烧残渣法、顶空气相色谱法对样品中的水分、灰分和溶剂残留进行检测,以扣除上述杂质对标准物质定值过程的影响。结果表明,标准物质中的水分含量为0.063%,灰分含量为0.082%,挥发性溶剂残留未检出。 因此,天麻素标准物质的定值结果为99.7%。 表4 天麻素标准物质纯度定值结果(n=6) 本研究中所制备的天麻素标准物质的不确定度主要是制备过程中不均匀性引起的、储存和运输过程中标准物质不稳定性造成的以及定值过程中引入的。 (1)制备过程中不均匀性引起的不确定度为: ubb=sbb=0.01%。 (2)该标准物质的稳定性考察时间为24个月,在储存和运输过程中存在的不确定度为: us=s(β1)×t=0.05%。 (3)定值时产生的不确定度包括:i.将合作实验室的平均值作为一组新的测量值,该组测量值的相对标准偏差作为不确定度来源之一,u1= 0.20%;ii.液相色谱响应因子引入的不确定度为u2=0.2%;iii.由于在定值方法研究过程中样品的进样量均在检测器的检测线性范围内,故仪器检出限引起的不确定度可忽略不计;iv.由于水分含量、灰分含量、溶剂残留的测定值均小于0.1%,此部分的不确定度可忽略不计。 扩展不确定度为UCRM=k×uCRM=0.6%,其中k=2。 本文采用半制备液相色谱系统从天麻提取液中分离纯化了天麻素,并以紫外光谱、红外光谱、质谱、核磁共振波谱等手段对分离纯化后的物质进行了定性分析,确定所制备的物质是天麻素。以高效液相色谱法结合面积归一化法对该标准物质的均匀性、稳定性进行了考察,考察结果经统计学计算,发现该标准物质的均匀性较好,同时该标准物质在二年内稳定。最后经国内8家资质合格的实验室进行联合定值,该标准物质的纯度为99.7%,扩展不确定度为0.6%(k=2)。所建立的天麻素标准物质制备方法为从中草药提取物中制备相关天然产物标准物质提供了一种完备的策略,同时该标准物质有助于满足含有天麻的相关食品、保健品分析检测方法校正以及质量控制工作需求,为用户在不同时间、不同空间,使用不同包装的该种标准物质时所得检测结果的准确性、可比性和溯源性提供保证。
2.4 稳定性检验


2.5 定值结果

2.6 不确定度结果
3 结论
