Cel48F产物排出过程中酶-底物相互作用及关键残基的分子动力学模拟
2019-02-22刘天志鄂镜雯
刘天志, 董 莹, 鄂镜雯, 韩 菲, 李 卓*, 张 浩
(1. 盛实百草药业有限公司, 天津 300000;2. 吉林大学 a. 理论化学研究所, 吉林 长春 130061, b. 白求恩制药厂, 吉林 长春 130012)
作为高等植物细胞壁的主要成分,纤维素广泛存在于植物和藻类等生物质中,是地球上分布最广、最丰富的可再生资源[1].纤维素可以被纤维素酶高效水解,先降解为短链糊精或纤维二糖,最终被降解为葡萄糖[2-3].这些单糖可以通过微生物发酵技术生产出生物乙醇,缓解因使用化石燃料带来的环境污染[4-7].
纤维素酶Cel48F是一种典型的具有连续内切活性的纤维二糖水解酶[8-9].晶体结构研究表明,Cel48F结构中有2个重要的区域[9,11]:底部是一个由6个α-螺旋围成的特定供水通道[12];上部是1个底物结合区域,该结合区域由1个底物结合通道和1个产物裂缝组成,特定供水通道、底物结合通道和产物裂缝三者形成1个Y字形结构[9].底物结合通道里有7个特定的糖单元结合子位置[13],分别为-7~-1,裂缝与通道相连,有4个特定的糖单元结合子位置+1~+4,水解发生在-1和+1子位置之间,在裂缝的4个子位置中,+1和+2是产物的初始结合位置,而+3和+4是虚位置,在野生型酶的实际水解过程中,底物链在水解前不可能达到这两个位置(因为水解产物是纤维二糖,因此水解前只可能占据+1和+2子位置),只是在产物排出过程中可能起到结合产物的作用[9,13].
在之前的研究中,笔者深入探究了纤维素酶过程性水解中的选择机制[14].另外,还对一个能显著影响酶活性但其具体作用尚不清楚的Glu 44在水解中的作用进行了详细研究,确定了Glu 44的一些可能作用,在研究Glu 44的作用过程中,使用了拉伸分子动力学模拟(SMD)的方法,发现Glu 44在产物纤维二糖从酶中排出过程中参与了氢键网络的重组,进而辅助底物排出[15].在进行该工作时,发现对该酶的产物解离过程目前尚没有完整的研究,之前进行的拉伸动力学研究虽然能考察产物的动态解离过程,但由于其需要固定的拉伸方向,以及拉伸过程中纤维二糖不可旋转等特点,导致其并不能完全真实地体现产物解离过程.想研究产物解离过程,最直接的办法是将底物在水解位置断开后,进行一个长的无任何限制的分子动力学模拟,直到最终产物从酶中脱离.但是,模拟该过程所需时间较长,在现有的计算条件下很难实现.好在晶体结构提供了产物解离过程中可能经过的特定结合位置(即+1~+4子位置),因此,可以手动将产物分别放在不同的结合位置上,再分别进行分子动力学模拟,研究产物在不同位置时的结合模式,最终就能汇总出大致的产物解离过程.
1 实验部分
1.1 初始构象获得
共模拟了4个酶-五糖复合物体系,4个体系的蛋白完全相同,其初始结构均来自蛋白质数据库(Protein Data Bank, PDB,编号1FAE).在4个体系的初始复合物中,底物和产物分别被连接在不同的位置,其中初始复合物A中底物为纤维五糖,结合于-3~+2子位置,即水解未发生前的酶-底物复合物;初始复合物B中糖单元也结合于相同的-3~+2子位置,但在-1和+1间糖苷键被手动断开,其中占据-3~-1子位置的是残余底物,占据+1~+2位置的为产物纤维二糖,这是水解发生后产物尚未移动时的复合物;初始复合物C和D中残余底物位置不变,产物分别结合于+2~+3和+3~+4子位置,为产物向出口连续移动1个和2个子位置时的复合物(见图1).
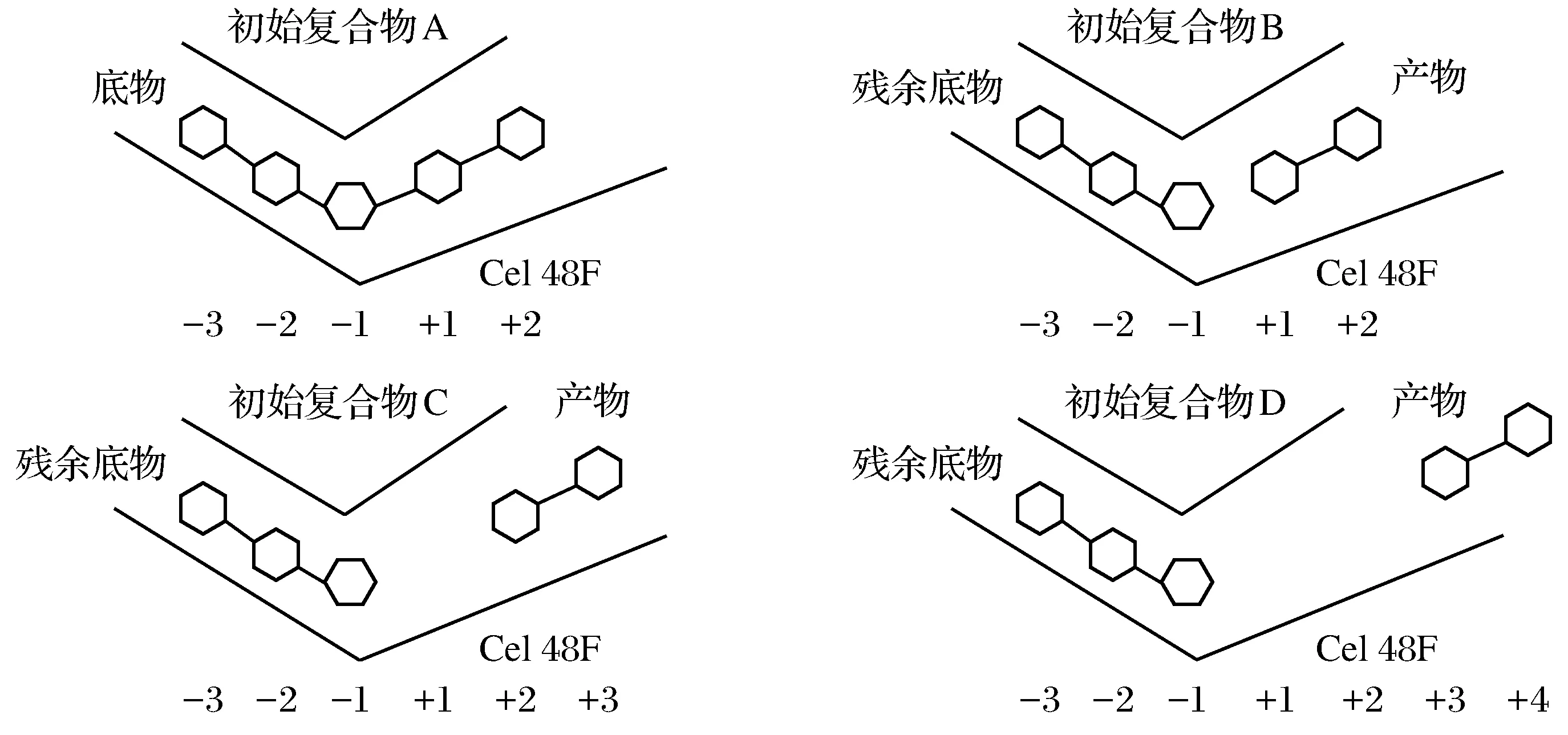
图1 本文模拟的4种初始复合物中底物和产物的位置Fig.1 The positions of substrate and product in all four initial complexes simulated in the present work
1.2 计算方法
(1) 分子动力学模拟.选用GROMACS 4.5.2分子动力学软件来研究生物分子体系[16],对Cel48F的蛋白分子和糖链底物分别施加CHARMM27力场和CHARMM22力场来生成各自的拓扑文件和力场参数文件[17].将体系置于正方体的水盒子中心,溶剂选取TIP3P水分子模型[18]并保持截断半径为1.0 nm.首先,用最陡下降法对复合物进行初步的结构优化来消除分子间不合理的构象碰撞.经过能量最小化之后,体系初步达到一个局部的能量低点.然后经过500 ns的NVT和500 ns的NPT过程,使体系最终达到恒定的温度(310 K)和1个标准大气压,其中长程静电力采取Particle Mesh Ewald(PME)的方法处理[19].压力和温度分别采用V-rescale和Parrinello-Rahman的方法进行拟合,而所有的成键原子间的距离通过LINCS算法来固定[20].最后,分子动力学模拟过程施加到整个体系中,积分步长为2 fs,轨迹每10 fs保存一次用于后续的数据分析.
(2) 结合自由能计算.蛋白质分子和配体之间的分解自由能通过Amber 10软件包的MM/PBSA程序来计算[21-22].在Amber ff99SB的立场下,100帧的构象从动力学轨迹中被提取出用来计算平均结合自由能.每个残基对配体的能量贡献会从三个方面被统计,包括静电作用(Gele),范德华作用(Gvdw),以及溶剂化作用(Gsolv),这三者中,静电作用经常与氢键相关,范德华作用经常与疏水相互作用相关.依据公式如下:总的残基-配体结合自由能等于这三项之和,表示为ΔGtotal=ΔEvdw+ΔEele+ΔGsolv[23].其中,溶剂化作用又分为极性溶剂化作用和非极性溶剂化作用.溶剂化自由能的计算采用Adaptive Poisson-Boltzmann Solver(APBS) 程序来计算.自由能的每一项贡献都取自于相同的构象计算.根据不同残基和配体的结合自由能强弱,可以了解在活性中心处对配体稳定有突出贡献的残基[24].
2 结果与讨论
2.1 总体构象对比
在分别以4个复合物A、B、C、D为初始构象进行了4个50 ns的分子动力学模拟后(所有4个模拟结果的三维结构可在支持信息中名为A.pdb,B.pdb,C.pdb,D.pdb的4个文件中找到),首先分析了4个模拟过程中均方根偏差(RMSD)的变化,除了复合物C是在约20 ns后达到平衡外,其余3个RMSD值都是在初始很短阶段的上升后(<1 ns)即达到平衡,平衡后的RMSD值在很小范围内波动(<0.05 nm).然后,将50 ns模拟后的4个构象,分别去掉底物,使用DS 2.5程序进行相似性对比,对比结果表明,以复合物A模拟后的构象为参考,复合物B、C、D的构象与A构象的相似性分别为0.819 844,0.824 882和0.819 156(相似性在0~1之间,越接近1说明其相似度越大),4个复合物蛋白部分相似性很高,说明产物的逐步排出并未引起蛋白构象的显著变化.
值得注意的是,在A、B、C构象中,无论底物是否在水解位置断开,在50 ns模拟之后,底物和产物的位置虽然均有变化,但都没有从蛋白中解离,而在D构象中,产物很快就从酶中解离(<10 ns).产物在不同构象中的这种差异是由于在Cel48F中,产物解离的主要动力是来自外界溶剂的作用,即当产物由亲脂的长链纤维素解离为亲水的纤维二糖之后,溶剂水分子容易将其包裹并带出酶中,实现产物解离[25].但是,由于Cel48F产物位置是逐渐变宽的裂缝,使得在初始解离和移动后(即产物在B、C两构象时),裂缝比较狭窄,并且裂缝侧壁上有很多疏水残基,使得外界水分子不容易进入到产物周围,因此产物也就不容易在短时间内被带出,而当产物到达D构象位置时,由于裂缝变宽,水分子容易进入并包裹产物,因此可在极短的时间内被带出酶中.
如上所述,由于D构象可以被认为是产物排出过程的终点,此位置产物与酶的相互作用很少,非常容易解离,与笔者研究的主题,即产物排出过程中产物与周围残基的相互作用关联较小,因此,后文中将不再讨论该构象.
2.2 关键残基的构象及相互作用变化显示其在产物排出中的作用
2.2.1 底物非还原末端周围残基及相互作用
底物的-3端为非还原末端,而+2端为还原末端,首先讨论非还原端,即-3~-1糖单元(在B、C复合物中是残余底物)周围残基与底物间的相互作用.
(1) 氢键相互作用.对A、B、C三个模拟,在其构象平衡阶段随机选取10个快照截图,使用GROMACS程序对底物和蛋白间氢键进行自动搜索,程序默认氢键范围为X…H…Y,键角大于120°,并且2个重原子X与Y之间距离小于等于0.35 nm.所有3个模拟的全部30个截图中存在的氢键列于表1中,其中一些较重要的氢键在A构象中与底物关系如图2所示.对于表1中的氢键,在3种构象中各自截取5 000个快照截图,并分别统计表1中的氢键在所有5 000个快照截图中存在的比例,也列于表1中.

表1 底物周围的氢键以及该氢键在所有3个模拟过程中提取的5 000帧快照中的存在率
从表1中可以看出,在-3~-1子位置一端,由于其位置在不同构象中未发生变化,因此酶中残基与其形成的氢键的变化也相对较小,在A、B构象中,存在几率最高的氢键几乎完全一致,包括Ser 54~-1糖单元,Glu 55~-1、+1糖单元的多氢键,Trp 298~-2糖单元,Trp 154~-3糖单元,Phe 180~-3糖单元.结合自由能的结果可以进一步验证这些氢键,表2~表4中列出了3种构象中拥有较明显静电、范德华或总结合自由能贡献的残基(绝对值大于4.182 kJ·mol-1).从A构象的结合自由能计算(表2)中可以看出,Trp 154和Phe 180中,并没有较明显的静电贡献,反而是范德华作用非常明显,这说明以上2个残基与底物间的靠近主要是由于形成疏水相互作用而不是氢键,高的氢键存在率只是因为在统计时相关原子恰好处于氢键范围内.不管是氢键还是疏水相互作用,在A、B构象中是相当稳定的,它们可以将底物束缚在特定的底物结合位置上.在这2种构象中,只有Asp 230~-1糖单元间的氢键存在率发生了较大变化(从99.84%降到9.68%),这可能与Asp 230的功能与其他残基不同有关,在之前的研究中,Asp 230被认为是关键结合亲核水分子的碱残基,因此,在水解前(A构象),它应与底物有较强的相互作用,以保证水分子能够进攻正确的位置,而在水解后(B构象),它应该远离底物,使逆反应不易发生.与A、B构象中糖单元位置基本没有变化不同,在C构象中,由于产物移动到了+2~+3子位置上,使其离残余的-3~-1糖链较远,两者间相互作用减小,因此导致了-3~-1糖链也有一个很小的位置变化并使其与底物间氢键有一定变化.但是,存在率最高的几个氢键,如Phe 180~-3糖单元、Trp 298~-2糖单元间的氢键仍能保持.氢键的变化主要体现在Glu 54~-1糖单元、Glu 55~-1糖单元、Trp 154~-3糖单元间氢键存在率的降低及新的Asn 227~-2糖单元和Tyr 323~-1糖单元间的氢键存在率的升高.

表2 A构象中有显著的结合自由能(绝对值大于4.182 kJ·mol-1) 的残基及其分解的结合自由能数值

表3 B构象中有显著的结合自由能(绝对值大于4.182 kJ·mol-1)的残基及其分解的结合自由能数值

表4 C构象中有显著的结合自由能(绝对值大于4.182 kJ·mol-1)的残基及其分解的结合自由能数值

图2 构象A中未水解的底物和酶中残基间的相互作用Fig.2 The interactions between unhydrolyzing substrate and residues in A conformation
(2) 疏水相互作用.除了氢键作用外,疏水相互作用在蛋白与底物结合过程中也起到重要作用.从表2中的结合自由能可以看出,在构象A中,在-3~-1一侧, Glu 55、Trp 154、Phe 180和Trp 298拥有最明显的范德华贡献(∣ΔEvdw∣>8 kJ·mol-1),并且这些范德华贡献在B和C构象中变化也不大(见表3,表4).
综上所述,-3~-1端底物构象及于蛋白相互作用的变化均不大,并且与笔者主要关心的产物排出无关,因此对这一侧不做重点讨论.
2.2.2 底物还原末端周围残基及相互作用
图3显示了将A、B和B、C构象分别进行对比的结果,结合氢键存在率及结合自由能的计算,就可以得到从水解开始到产物排出过程中产物和关键残基的变化.首先对比A和B构象(如图3a所示),在+1~+2子位置间,A与B构象中存在的氢键的最明显差别在于,A构象中,Glu 55主要与+1糖形成氢键,Glu 44与+2糖单元间氢键存在率较低(67.93%),另外,Trp 611与+1糖单元间也有一定的氢键存在率(53.71%).尽管Glu 44、Trp 611在此构象中与底物间形成的氢键并不稳定,存在率只略高于50%,但是A构象中的结合自由能显示,此时这2个残基与底物之间,都已经有比较明显的静电相互作用(静电贡献分别为-11.62 kJ·mol-1和-6.02 kJ·mol-1).而B构象中,由于-1与+1间糖苷键断开,产物纤维二糖不再受到-3~-1底物的限制,因此它在Glu 44和Trp 611的作用下,向Glu 44方向滑动了一小段距离,使得原本只是有相互作用但还未形成稳定氢键的Glu 44~+2糖单元,Trp 611~+1糖单元间均形成了稳定的氢键(氢键存在率上升到94.5%和74.91%),同时,表3中这2个残基的静电贡献也大幅增强(分别达到-27.54 kJ·mol-1和-10.91 kJ·mol-1).该滑动还使Glu 55与+1糖单元间距离变远,并进一步导致两者间的氢键断开,Glu 55转而与-1糖单元形成氢键(尽管表1中A、B两种构象里Glu 55与底物的氢键存在率相差不大,但自动分析不能区分其到底与哪个糖单元形成氢键,更详细的构象分析表明在A中,Glu 55主要与+1糖形成氢键,而在B中,其主要与-1糖形成氢键).这个小的移动可能是Glu 44(或者也包括Trp 611)在酶催化水解过程中最关键的作用,它使产物离开残余底物和关键催化残基(Glu 55被认为是催化中对活性影响最大的提供质子的酸残基[3,26])一段距离,避免了逆反应的发生,相当于加快了水解反应的速度,这可以解释实验中将Glu 44突变为Gln 44后,酶活性显著降低却没有完全消失的现象.

图3 分别对比(a)A/B构象间和(b)B/C构象间与产物发生相互作用的残基
继续对比B和C构象(图3b)可以看出,当产物从B构象所处的位置继续向外滑动过程中,其内侧的糖单元(初始构象中位于+2子位置)明显向产物裂缝上方移动,并与该方向上的Thr 410形成氢键,而外侧糖单元(初始构象中位于+3子位置)则只是受内侧糖单元影响轻微地向上方发生位移.在B构象中,Thr 410与产物间的氢键存在率为0,而在C构象中,虽然该氢键的存在率仅为12.88%,但值得注意的是,由于模拟过程中,产物是逐渐向Thr 410方向移动的,因此,实际上在模拟40 ns以后,产物与Thr 410才进入氢键范围.而40~50 ns区间内的氢键统计显示,Thr 410与底物形成氢键的比例已高达50.2%.另外,结合自由能的计算也显示在C构象中,Thr 410与底物间的静电相互作用也有显著增强(从-0.38 kJ·mol-1变为-7.03 kJ·mol-1).
结合自由能计算能明显地显示出内侧糖单元向上移动的驱动力,从表2~表4中的结合自由能结果中可以看出,尽管A和B构象中,底物与裂缝上方残基都没有明显的静电作用,但其中一个拥有大的芳香环的Trp 411,在A、B、C三个构象中均显示出了非常显著的范德华贡献,可以认为,该残基是从B向C构象变化中使产物向裂缝上方移动的初始驱动力,而在产物移动到接近C构象的位置后,该方向上另外几个残基,包括Pro 406、Thr 410、Thr 462、Glu 542和Arg 544,与产物形成了较明显的相互作用,使产物进一步向它们靠近.在产物裂缝的上方,即Thr 410和Trp 411附近,裂缝开口较大,溶剂水分子更容易进入并最终带走产物.
对外侧糖单元而言,Asp 494与其形成的氢键存在率迅速上升(从B构象中39.03%到C构象中的62.28%),特别是在40 ns以后,其存在率超过了80%,表4中的结合自由能结果也显示Asp 494与底物间有很明显的静电相互作用(10.91 kJ·mol-1).正是由于Asp 494的存在,使得外侧糖单元并没有向上方移动太多.这种作用使产物大致体现出一种以外侧糖单元为中心的转动,导致其不再紧贴产物裂缝的一侧,并在内侧糖单元原本的位置上留下一个空腔,使外部的溶剂水分子容易进入并包裹产物.
新相互作用形成的同时,B构象中原有的Glu 44与Trp 611与底物形成的氢键存在率在C构象中都大幅下降,但是,结合自由能计算表明,尽管这2个残基与底物距离超出了正常氢键,但它们与底物间的静电贡献,虽然也有所减小,但仍十分明显,这种相互作用,可能使产物在C构象所处的位置附近上下震动,帮助溶剂水分子更容易进入产物内侧.
3 结 论
从以上模拟结果的讨论可知,由于-3~-1位置的糖单元没有明显变化,因此其在产物排出过程中与周围残基所形成的相互作用变化不大,特别是主要维持底物位置的Glu 55~-1或+1糖单元,Phe 180~-3糖单元,Trp 298~-2糖单元间的氢键,以及Glu 55、Trp 154、Phe 180和Trp 298等与底物间的疏水相互作用.而在+1~+2端,相互作用变化较大,产物先是在Glu 44和Trp 611作用下向出口移动一个小的距离,使产物远离残余底物,防止逆反应发生,接着,产物继续向外移动,在此过程中其内侧糖单元在Trp 411作用下向产物裂缝上方移动,并与Thr 410形成弱氢键.而外侧糖单元则与Asp 494形成氢键,并在该残基束缚下,只轻微向裂缝上方移动.内外侧糖单元不同运动模式,使产物整体发生转动,并在内侧糖单元原本的位置上留下一个空腔,使外部的溶剂水分子容易进入并包裹产物.
