谷氨酰胺转氨酶在Yarrowia lipolytica中的活性表达
2019-02-15任蕊蕊李江华堵国成
任蕊蕊,刘 松,李江华,堵国成,陈 坚
(1.江南大学 生物工程学院,无锡 214122;2.江南大学 工业生物技术教育部重点实验室,无锡 214122)
谷氨酰胺转氨酶(EC 2.3.2.13,Transglutaminase,TGase)可以催化肽链中的谷氨酰胺残基中的γ-羧酰胺基与酰基受体发生转酰基反应,从而使蛋白质或多肽之间发生共价交联[1]。基于特殊的催化反应,TGase可使蛋白质与赖氨酸交联,提升食品的营养价值[2];也可改善蛋白类食品功能的特性,如持水性、黏性、乳化稳定性,减少储藏损失和烹饪损失[3]。因此,TGase在食品加工领域有着广泛的应用。近年来,TGase在生物技术研究和医药[4-7]、纺织业[8]及皮革加工[9]等领域亦展现了较大的应用前景。
基于N-端酶原区对折叠与分泌的重要影响[10-11],TGase通常以无活性的酶原(pro-TGase)形式在异源宿主中表达,再经体外蛋白酶切除酶原区后才得到活性TGase[12-13]。显然,这种TGase表达策略不利于工业化生产。近年来异源表达活性TGase的策略主要有两种:pro-TGase与活化蛋白酶共表达;酶原与成熟TGase共表达。Kikuchi等[14]在谷氨酸棒杆菌(Corynebacteriumglutamicum)中共表达pro-TGase与蛋白酶SAM-P45,活性TGase产量达到142 mg/L。Yurimoto等[15]与李鹏飞等[16]分别在甲醇营养型酵母(Methylotrophicyeasts)和毕赤酵母(Pichiapastoris)中共表达,实现了活性TGase的分泌。
尽管研究者已成功获得TGase在C.glutamicum和P.pastoris等宿主菌中的活性表达,但总体产酶水平不高。解脂耶氏酵母(Yarrowialipolytica)为食品安全菌[17],发酵中不需诱导或添加抗生素,能用于食品和药品相关酶或蛋白的生产[18]。本研究拟以Y.lipolytica为宿主,通过共表达茂源链霉菌(Streptomycesmobaraensis)谷氨酰胺转氨酶激活金属蛋白酶(TAMEP)[12-13,19]及在TGase N-端插入宿主Kex2蛋白酶识别位点(KR)[20],实现活性TGase高效分泌表达。
1 材料与方法
1.1 材料
1.1.1 菌株与质粒
TGase基因来源菌株S.mobaraensis、质粒pINA1297/N355Q[21]、pINA1297、Y.lipolyticapo1h/1297[21]和宿主菌Y.lipolyticapo1h由江南大学生物系统与生物加工工程研究室保藏。pUC57/TAMEP由上海睿迪生物科技有限公司合成。
1.1.2 酶、试剂、引物和DNA序列测定
限制性核酸内切酶购自Thermo Fisher Scientific公司,胶回收试剂盒和Competent Cell Preparation Kit试剂盒购自Takara(大连)公司,Bradford蛋白浓度测定试剂盒购自碧云天公司。引物合成和DNA测序由生工生物工程(上海)股份有限公司完成。
1.1.3 培养基
种子培养基(YPD,g/L):酵母膏 10,蛋白胨 20,葡萄糖 20。
筛选培养基(酵母基础氮源培养基,YNB,g/L):YNB 6.7,葡萄糖20。
固体培养基则是在液体培养基中加2%的琼脂。
发酵培养基(g/L):甘油 15,酵母膏 20,氯化铵2.64,磷酸二氢钾 0.32,无水硫酸镁 0.25,维生素B13.34×10-4,调pH 8.0。
1.2 方法
1.2.1S.mobaraensis基因组的提取
用FastPrep破碎仪破碎菌体,破碎强度为6.5,破碎时间为20 s,每个样品破碎10次。然后用植物基因组提取试剂盒(天根生化科技有限公司,北京)提取基因组,具体操作按说明书进行。
1.2.2 质粒pINA1297/pro-TGase的构建
以pINA1297/N355Q为模板,P1和P2为引物进行PCR扩增含有酶原区pro的pINA1297表达载体。以基因组为模板,P3和P4为引物进行PCR扩增TGase基因片段。两种PCR产物经DpnI消化后进行胶回收,回收产物以摩尔比为1∶2进行混合,使用One Step Cloning Kit进行连接后,转化E.coliJM109,获得表达载体pINA1297/pro-TGase。
1.2.3 插入Kex2蛋白酶识别位点重组质粒pINA1297/pro-KR-TGase的构建
以质粒pINA1297/pro-TGase为模板,P5和P4引物进行PCR,扩增含有已加入Kex2蛋白酶识别位点(KR)[20]的茂源链霉菌TGase基因片段。PCR产物经DpnI消化后进行胶回收,再以回收产物作为引物,以质粒pINA1297/pro-TGase为模板进行大引物PCR,获得突变基因片段。PCR产物经DpnI消化后进行胶回收,转化E.coliJM109,获得重组质粒pINA1297/pro-KR-TGase。
1.2.4 活化蛋白酶重组质粒pINA1297/TAMEP的构建
以质粒pINA1297/N355Q为模板,P1和P2为引物进行PCR,通过PCR扩增含有酶原区的pINA1297表达载体。以pUC57/TAMEP为模板,P6和P7为引物进行PCR,通过PCR扩增TAMEP基因。两种PCR产物经DpnI消化后进行胶回收,回收产物以摩尔比为1∶2进行混合,使用One Step Cloning Kit进行连接后,转化E.coliJM109,获得重组质粒pINA1297/TAMEP。
1.2.5Y.lipolytica的转化与重组子的筛选
重组质粒经快切酶NotI酶切后,胶回收得到相应的线性化质粒。然后用醋酸锂转化法[22]转化Y.lipolyticapo1h感受态,质粒通过zata整合位点将突变基因整合到Y.lipolyticapo1h(Ura-,ΔAEP,ΔAXP,Suc+)基因组中。整合成功的Y.lipolyticapo1h会使其从尿嘧啶(ura)缺陷型菌株转化为非缺陷型菌株。通过缺乏ura的YNB培养基28℃,培养3~6 d后,获得重组菌的阳性转化子。
基于上述转化方法,将pINA1297/pro-TGase转化Y.lipolyticapo1h感受态获得重组Y.lipolyticapo1h/pro-TGase;将pINA1297/pro-KR-TGase转化Y.lipolyticapo1h感受态获得重组Y.lipolyticapo1h/pro-KR-TGase;将pINA1297/TAMEP转化Y.lipolyticapo1h/pro-TGase感受态获得重组Y.lipolyticapo1h/HM+HT。

表1 基因克隆及定点突变引物Table 1 Primers used for gene cloning and site-specific mutagenesis
1.2.6 重组Y.lipolytica的发酵
选取10~15个重组菌的阳性转化子,接种于YPD液体培养基中,于28℃,200 r/min的摇床中培养24 h。将种子液以10%的接种量转接于发酵培养基中,28℃,200 r/min摇瓶培养120 h。
1.2.7 TGase的酶活测定
采用比色法测定TGase酶活[23]。1个单位的酶活定义为:在37℃的条件下,每分钟催化α-N-CBZ-GLN-GLY[24](Sigma)合成1 μmol 的L-谷氨酸-γ-单轻胺酸所用的酶量(U/mL)。
1.2.8 菌体量的测定
利用紫外可见光分光光度计(UV2450,Shimadzu,日本)检测菌液在600 nm波长处的吸光度值,即OD600。
1.2.9 纯化方法
1.2.10 浓度的测定
蛋白质浓度测定采用Bradford蛋白浓度测定试剂盒,具体按说明书操作。
1.2.11 动力学常数的测定
在底物浓度为0~30 mg/mL范围内设定浓度梯度,均按1.2.6中所述的方法测酶活,通过双倒数作图法计算可得动力学参数Km值。
2 结果与分析
2.1 重组Y.lipolytica po1h/pro-KR-TGase的构建及摇瓶发酵
将S.mobaraensispro-TGase基因克隆至pINA297得到表达质粒pINA297/pro-TGase(图1),转化Y.lipolyticapo1h得到的重组菌Y.lipolyticapo1h/pro-TGase。发酵分析显示,Y.lipolyticapo1h/pro-TGase上清液检测到TGase活力仅为0.13 U/mL,但在49 ku左右具有明显的pro-TGase电泳条带(图2),大于理论值43 ku,可能是由于酶表达时发生了糖基化[21]。上述结果表明,pro-TGase在Y.lipolytica中能高效表达,但不能高效转化为活性TGase。
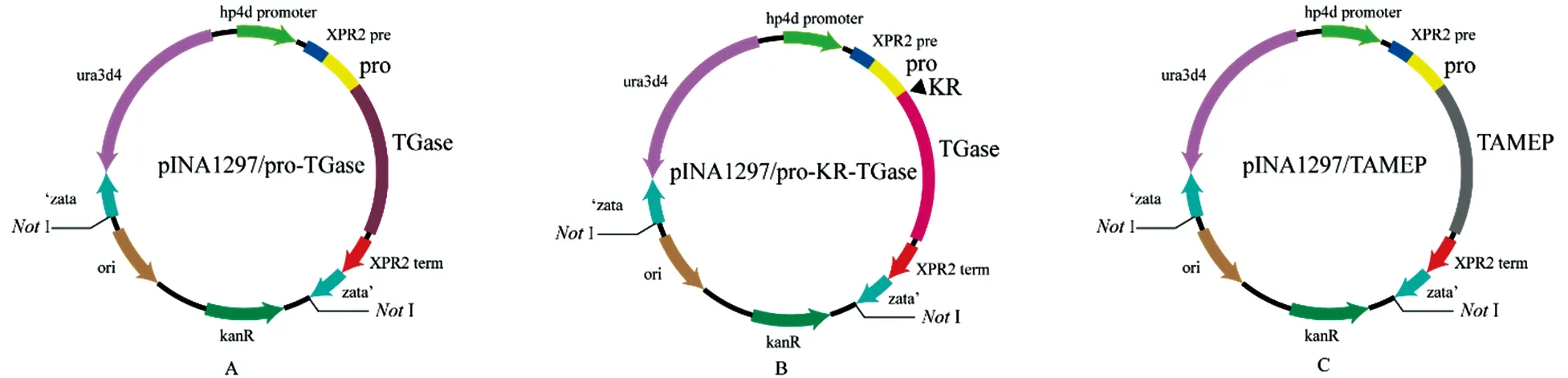
A:pINA297/pro-TGase;B:pINA297/pro-KR-TGase;C:pINA297/TAMEP
图1重组质粒示意图
Figure 1 The plasmids constructed in this study
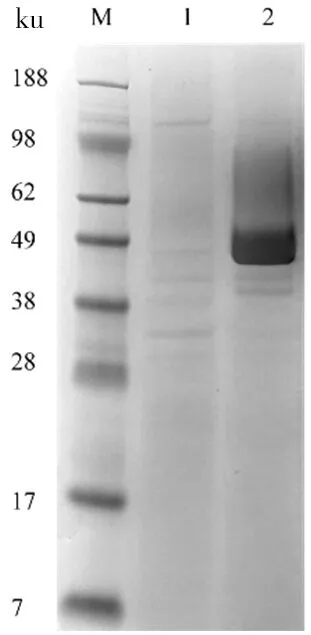
M:Marker;1:Y.lipolyticapo1h/1297;2:Y.lipolyticapo1h/ pro-TGase
图2Y.lipolyticapo1h/pro-TGase生产TGase SDS-PAGE分析图
Figure 2 The SDS-PAGE analysis of TGase produced byY.lipolyticapo1h/pro-TGase
Kex2蛋白酶属于亚麻酶家族,该家族的酶识别位点专一,具有将酶原区加工成有活性酶的功能[25]。Richard等[26]发现了Kex2蛋白酶也存在于Y.lipolytica中。因此,本研究采用在酶原区与TGase之间加入Kex2蛋白酶的识别位点KR[20]的策略,构建了质粒表达载体pINA1297/pro-KR-TGase(图1-B),确认pro-KR-TGase基因序列与公布的序列完全一致。质粒经NotI线性化后转化Y.lipolyticapo1h感受态获得重组Y.lipolyticapo1h/pro-KR-TGase。酶活测定和SDS-PAGE检测发现:发酵0~20 h酶活变化很小,几乎全部以非活性酶原的形式表达;20~76 h,TGase的活性基本呈直线上升,最高酶活达5.26 U/mL;76~120 h后,TGase持续降解,活性基本呈直线下降(图3-A、B)。结果表明,添加Kex2蛋白酶识别位点明显促进了pro-TGase转化为活性TGase的效率。
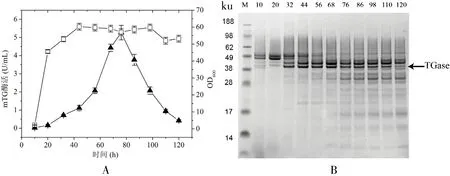

图3重组菌Y.lipolyticapo1h/ pro-KR-TGase发酵生产TGase的活性(A)及SDS-PAGE分析(B)
Figure 3 The production of TGase byY.lipolyticapo1h/ pro-KR-TGase in shake flask(A),and its SDS-PAGE analysis(B)
2.2 重组菌Y.lipolytica po1h/HM+HT的构建和摇瓶发酵
TAMEP蛋白酶最初从S.mobaraensis中分离出来,可以识别酶原区pro序列C-端的FRAP,从而可以切除酶原区,使pro-TGase转化为活性TGase[19]。由于Y.lipolytica中没有TAMEP,本研究首次采用共表达pro-TGase与TAMEP的方法,构建了重组Y.lipolytica/HM+HT。重组菌摇瓶发酵,酶活测定和SDS-PAGE分析结果显示,重组菌Y.lipolyticapo1h/HM+HT发酵 0~20 h酶活变化很小,几乎全部以非活性酶原的形式表达;20~110 h,TGase活性基本呈线性缓慢增长,并达到最大值6.77 U/mL(图4-A、B);110~120 h,TGase酶活有下降趋势。结果表明,添加共表达TAMEP明显促进了pro-TGase转化为活性TGase的效率。
2.3 酶反应动力学分析
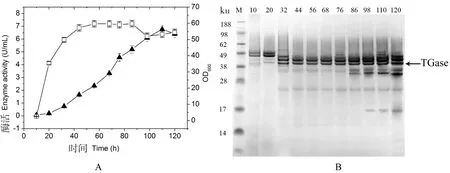
图4重组菌Y.lipolyticapo1h/ HM+HT发酵生产TGase的活性(A)及SDS-PAGE分析(B)
Figure 4 The production of TGase byY.lipolyticapo1h/HM+HT in shake flask(A),and its SDS-PAGE analysis (B)
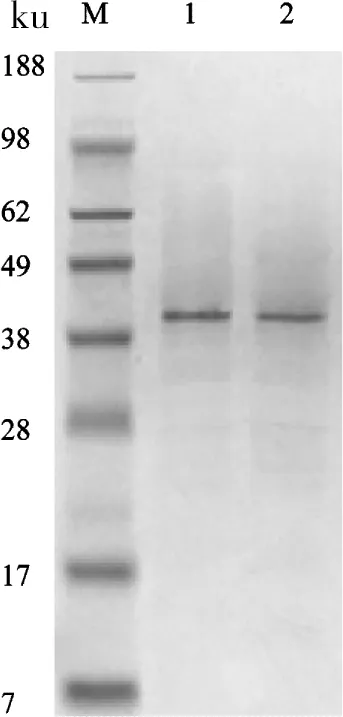
M:Marker;1:pro-KR-TGase;2:HM+HT
图5 pro-KR-TGase及HM+HT纯化后SDS-PAGE分析
Figure 5 The SDS-PAGE analysis of pro-KR-TGase and HM+HT
糖基化对酶的正确折叠、分泌、酶活力、酶亲和力、底物特异性等具有重要影响[27]。王小燕等[28]通过对β-葡萄糖醛酸苷酶N115和N418进行糖基化修饰提高了酶对底物的亲和力,对N26和N150进行糖基化修饰提高了酶的催化效率。pro-TGase在Y.lipolytica中表达时会发生糖基化[21],而S.mobaraensisTGase未发生糖基化。因此,糖基化可能是导致本研究中TGase的比酶活、酶与底物亲和力和催化活力等性质优于S.mobaraensisTGase的重要原因。
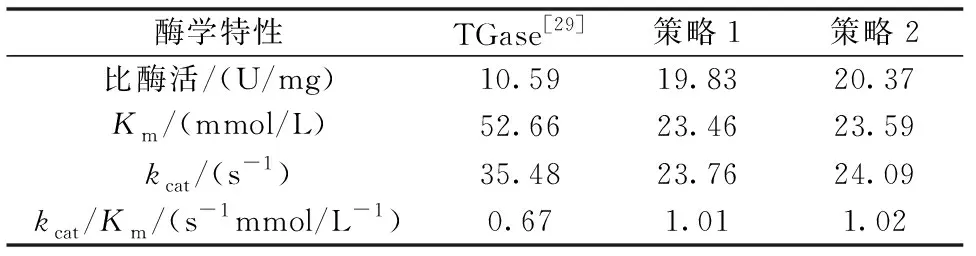
表2 TGase及其突变体酶学特性Table 2 Enzyme properties of TGase and its mutants
3 讨论
为促进活性TGase的异源表达,研究者开展了大量的工作。Kikuchi等[14]在C.glutamicum中共表达pro-TGase与蛋白酶SAM-P45,活性TGase产量达到142 mg/L。Yurimoto等[15]在M.yeasts中将pro和TGase作为两个独立的元件进行共表达,得到的TGase产量为1.83 U/mL;李鹏飞等[16]利用同样的方法在P.pastoris中实现了活性TGase的分泌表达,摇瓶产量最高为0.25 U/mL。本研究通过在pro与TGase直接插入Kex2识别位点KR和共表达pro-TGase与活化蛋白酶TAMEP两种策略,均获得了活性TGase的分泌,摇瓶发酵酶活分别可达5.26 U/mL和6.77 U/mL。与其它报道相比,本研究获得的活性TGase具有产量优势。此外,与S.mobaraensis成熟TGase相比,通过以上两种策略得到的成熟TGase的比酶活;酶与底物的亲和力Km及酶的催化效率kcat/Km等酶学性质明显提高。为TGase的工业化生产提供了新型高产菌种。
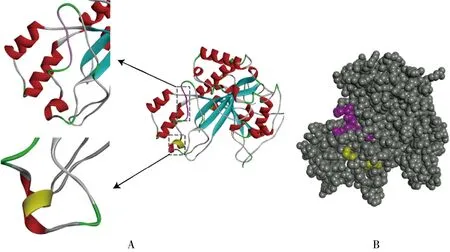
图6 谷氨酰胺转氨酶TGase的3-D结构Figure 6 Analysis on 3-D structure of TGase
黄色和紫色分别代表TGase的KR和FRPAP所在TGase 3-D结构的位置(yellow part represents the position of the amino acid sequence Lys-Arg in the TGase 3-D structure,and the purple part represents the location of Phe-Arg-Pro-Ala-Pro)
采用策略1构建的重组菌Y.lipolytica/pro-KR-TGase,在发酵76 h时酶活达到最高,但在发酵76 h之后存在成熟TGase持续降解的现象。其可能的原因是:S.mobaraensisTGase的第214~215位氨基酸序列为KR,位于一段较短的α-螺旋上,且位于酶蛋白表面(图6),可以被Kex2蛋白酶识别并切割,导致成熟TGase降解。采用策略2构建的重组菌Y.lipolytica/HM+HT,在发酵110 h时酶活达到最高,但在发酵86 h之后也存在成熟TGase持续降解的现象。其可能的原因是:成熟区TGase第223~227的氨基酸序列为FRPAP,位于TGase的一段loop环上,也位于酶蛋白表面(图6),且该序列与TAMEP蛋白酶的识别位点相似,易于被TAMEP识别并切割,进而导致成熟TGase降解。在后续研究中,将进一步鉴定TGase内部的Kex2和TAMEP蛋白酶识别位点,抑制发酵后期活性TGase的持续降解。
