弯枝藻属rbcL基因的适应性进化分析
2019-02-13韩雨昕南芳茹巩超彦冯佳吕俊平刘琪谢树莲
韩雨昕, 南芳茹, 巩超彦, 冯佳, 吕俊平, 刘琪, 谢树莲
弯枝藻属L基因的适应性进化分析
韩雨昕, 南芳茹, 巩超彦, 冯佳, 吕俊平, 刘琪, 谢树莲*
(山西大学生命科学学院,太原 030006)
为探讨淡水红藻的叶绿体基因及其适应性进化特征,选取弯枝藻属()及相近外类群的L基因共17条,利用PAML 4.9软件,对弯枝藻属L基因编码蛋白进行生物信息学分析,并分别采用分支模型、位点模型以及分支-位点模型对基因的选择位点进行检测。结果表明,弯枝藻属L基因编码蛋白的二级结构主要由螺旋和折叠构成,结构稳定。采用最大似然法构建的系统发育树表明,内类群为单一物种,分为3个小分支,具有一定地理分布规律。在3种进化模型中均未检测到统计上显著的正选择位点,表明绝大多数位点处于负选择压力下。因此,弯枝藻属L基因未发生适应性进化。
弯枝藻属;L基因;蛋白质结构预测;适应性进化
红藻主要分布于海洋中,淡水分布的红藻只占一小部分,有研究表明, 淡水红藻是海洋红藻在海陆变迁过程中遗留于淡水中,并在封闭环境中经过长时间演化而产生的一个重要类群[1]。淡水红藻大部分以固着方式生活在温度较低的清洁水体中的岩石或其他物体上,生存环境相对稳定和封闭[2–3]。弯枝藻属()是淡水红藻中的典型类群之一,早在190多年前就有记载,在世界上分布广泛,北美、加勒比群岛、西大西洋、亚洲、澳大利亚和夏威夷群岛等均有分布[4]。Necchi等[5]认为该属不同种样本间的遗传多样性较低,为一全球单种属。
1,5-二磷酸核酮糖羧化/加氧酶(Rubisco, E.C.4.1.1.39)存在于植物的叶绿体基质中,是参与植物光合作用的关键酶,约占可溶性蛋白质总量的50%[6]。它是一个双功能酶,既能催化RuBP与CO2反应生成3-磷酸甘油酸的羧化反应,又能在光呼吸中催化RuBP与O2反应氧化裂解形成 3-磷酸甘油酸、磷酸和磷酸乙醇酸,因此Rubisco对净光合率具有决定性的影响[7–8]。Rubisco固定CO2的活性位点位于大亚基,是由来自叶绿体基因组的L基因编码[9]。不同种类的绿色植物中Rubisco活性有较大差异,环境压力的限制可导致编码Rubisco大亚基的L基因发生适应性进化。在很多陆生植物L基因中检测出正选择位点,在藻类植物如串珠藻目L基因中也检测出有少数正选择位点[10]。
对具有重要功能的蛋白质进行适应性进化分析,有助于我们更加深入地了解当面对环境压力时,一些氨基酸的结构和功能会发生哪些改变[11]。目前对高等植物的适应性进化研究较多。周媛等[12]对凤尾蕨科(Pteridaceae)旱生蕨类的L基因进行了适应性进化研究,检测出多个正选择位点,其中有3个位点对维持Rubisco功能起重要作用。张丽君等[13]对蕨类植物的4基因进行了适应性进化研究,但未检测出正选择位点,表明该基因结构与功能已趋于稳定。此外还对黄花蒿()植物L基因[14]、麻黄科(Ephedraceae)植物L基因[15]和稻属() AA型物种叶绿体基因组[16]等进行了适应性进化研究。然而目前对藻类植物的适应性进化研究仍然很少。
作为全球分布的淡水红藻中的一个重要类群,弯枝藻属是如何适应不同的环境压力,在这些环境压力下,重要蛋白Rubisco大亚基是否发生了适应性进化?本研究分析了该属L基因的适应性进化,以期为探究其在海陆变迁过程对淡水环境的适应研究提供参考。
1 材料和方法
1.1 序列数据采集及系统发育树构建
从GenBank数据库中下载弯枝藻属以及外类群的L基因序列,共获得17条序列(表1)。以Clustal X软件[17]对序列进行对位排列,并进行人工校对,每条序列均包含350个密码子,以此作为构建系统发育树的基础。
采用MEGA 7.0软件[18]对序列特征进行分析, 应用Modeltest 3.7软件[19]对联配结果进行模型选择,选取的最佳核苷酸进化模型为TIM+I+G,其中I为进化速率恒定位点的比率,G为Gamma密度函数,K为估算参数的数目(表2)。运行PhyML 3.0软件[20],采用最大似然法(ML)[21]构建系统发育树。
1.2 生物信息学分析
选取登陆号为JX028169的L基因序列作为参考序列,以氨基酸序列的形式上传Prot Param (http://web.expasy.org/protparam/),对所选弯枝藻属L基因编码蛋白质的理化性质进行分析,并利用ProtScale (http://web.expasy.org/protscale/)预测该蛋白质的亲水性/疏水性[14]。
利用NetPhos 3.1 Server (http://www.cbs.dtu.dk/ services/NetPhos/)对蛋白质进行磷酸化位点预测。

表1 用于本研究的rbcL基因GenBank登录号

表2 Modeltest 3.7检验得到的rbcL基因优化模型参数
利用软件包DNASTAR.Lasergene. V 7.1中Protean模块对该蛋白质的二级结构进行分析[22–23]。
1.3 适应性进化分析
在适应性进化研究中,可以用核苷酸的非同义替换率(dN)与同义替换率(dS)的比值()来度量蛋白质水平上的选择压力,并进一步判断选择压力在非同义替换的固定过程中起阻碍或推动作用。一些核苷酸的替换不会引起氨基酸改变,称为同义替换;而更多情况下密码子的替换会引起氨基酸改变,称为非同义替换[24]。=1,即dNdS,表明选择对适合度无影响;<1,即dN
以最大似然法(ML)构建的系统发育树为基础数据,通过PAML 4.9软件包[27]中的codeml模块, 分别采用分支模型、位点模型和分支-位点模型进行适应性进化分析。
分支模型[28]中,允许非同义替换率和同义替换率的比值在不同分支上有变化。其中单一比率模型最为简单,该模型假定在所有进化支上值均相同;自由比率则设定各分支的值各不同。此外, 本研究也采用了二比率模型来进行检测。
位点模型[29]中,假定不同位点存在不同的选择压力,即值不同,但在系统发育树的不同分支中无差异。这一模型主要用于检测L基因是否存在正选择(>1)和负选择(<1)位点。本研究中采用的三对比较模型分别为:M1a (近中性)和M2a (选择)、M0 (单一比值)和M3 (离散)、M7 (beta)和M8 (beta &),前者为零假设,后者为备择假设。M0 (单一比值)对M3 (离散)模型检测各位点是否存在不同的值,并不检测正选择位点。对3对模型进行LRT检验(likelihood ratio test),通过比较模型间差异的显著性来检验正选择位点,在相对自由度(两模型参数数目之差)下,运用2分布进行显著性检验。
分支-位点模型[30]中,将系统发育树分为前景支和背景支,仅允许前景支中出现正选择位点及分支,对其进行LRT检验,在test1中将MA和M1a进行似然比检验,在test2中将MA和无效模型(设置为1)进行比较。经研究比较,选择test2算法更为可靠。
2 结果和分析
2.1 系统发育树构建
从利用最大似然法(ML)构建的系统发育树(图1)可见,内类群由单一物种弯枝藻()组成,并分为3个小分支,分支间存在明显的地域分布特点,A分支样本产地主要为中国和澳大利亚,B分支为北美(后验概率达81.1%),C分支为马来西亚、印尼及太平洋岛群等(后验概率为65.6%)。据此选取A、B、C共3个分支进行后续分析。
2.2 生物信息学分析
Rubisco大亚基的疏水/亲水性预测 衡量蛋白质亲水性/疏水性是根据GRAVY值,正值为疏水性蛋白质,负值为亲水性蛋白质。利用Prot Param测得Rubisco大亚基的GRAVY值为-0.099,表明具有亲水性,推断为水溶性蛋白质。从图2可知,弯枝藻Rubisco大亚基氨基酸序列在第190位异亮氨酸的GRAVY最高(3.211),表明该位点具有极强疏水性;第224位的GRAVY最小(-2.500),表明该位点具有极强亲水性[14]。
Rubisco大亚基磷酸化位点预测 用NetPhos3.1 Server对Rubisco大亚基磷酸化位点进行预测(图3), 结果表明,丝氨酸(Ser)磷酸化位点有9个,分别位于37、106、133、154、156、203、223、226和294位点,苏氨酸(Thr)磷酸化位点有6个,分别位于98、157、171、224、254和272位点,酪氨酸(Tyr)磷酸化位点有3个,分别位于5、164和176。
Rubisco大亚基的二级结构及保守结构域预测采用DNAStar软件包中Protean模块,对Rubisco大亚基的二级结构进行预测(图4), 采用Garnier-Robson方法计算特定氨基酸残基在特定结构内部的可能性,Chou-Fasman方法通过序列氨基酸残基的晶体结构预测蛋白质的二级结构,两种方法预测的蛋白质二级结构存在差异。Garnier-Robson方法预测弯枝藻Rubisco大亚基有19个螺旋,24个折叠,12个转角以及一些小片段的无规则卷曲。Chou- Fasman方法则预测有15个螺旋,10个折叠, 19个转角。两种方法预测的螺旋分别位于第1~6位、第26~38位和第49~67位,折叠分别位于第17~ 25位、第41~49位和第78~82位,转角结构位于第13~15位、第73~75位和第85~88位,Garnier- Robson方法预测的无规则卷曲分别位于第12~13位、第71~72位和第76~77位。
2.3 适应性进化分析
表3和4为各模型选择位点的鉴定结果。分支模型中,二比率模型指定分支A、B、C为前景支,其他为背景支。各前景支的估计值均小于1,表明各分支均处于负选择压力下。自由比率模型检测大多数分支值远小于1,仅有两个小分支(序列登录号为JX028153、KR706528)的值为999.0,因此对这两个分支进行了分支-位点模型检测。对分支模型中二比率模型A、B、C以及自由比率模型进行LRT检验(表4),其中分支B的结果较为可靠(< 0.05),其余分支后验概率表明均不具有可靠性。
位点模型中,模型M3 (离散)、M2a (选择)和M8 (beta &)允许>1,与其对应的零假设模型为M1a (近中性)模型、M0 (单一比值)模型和M7 (beta)模型。M3模型显著优于M0零假设模型(<0.01),表明各位点间承受的选择压力具有差异性。M2a (选择)模型中检测到1个正选择位点170Q (后验概率为51.3%),M8 (beta &)模型中检测到170Q (后验概率为61.1%)和180I (后验概率为60.3%)为正选择位点,但经LRT检验,拒绝存在正选择位点的假设(表4)。
分支-位点模型中,指定5个分支为前景支,其中分支D的序列登陆号为JX028153,分支E的序列登录号为KR706528。分支C和分支E没有检测到正选择位点。在分支A检测出98L (后验概率为68.6%)和327Q (后验概率为69.8%),分支B检测出217R (后验概率为89.3%)、225F (后验概率为89.4%)和309D (后验概率为89.8%),分支D检测出327Q (后验概率为94.0%)等为正选择位点,但似然比检验拒绝存在正选择位点的假设(表4),因此分支A、B和D的检验不能作为可靠的正选择位点证据。
3 讨论
研究表明,弯枝藻属L基因中未检测到正选择位点,说明其在进化过程中经受了严重的负选择。L是一个十分古老的基因,广泛存在于几乎所有高等和低等植物叶绿体中。因此在漫长的进化过程中,L基因很可能在结构和功能上已经趋于稳定,其适应性进化有可能在早期(数百万年前)已被固定,后来正选择位点被大量积累的中性替换位点或净化作用所掩盖,最终使正选择位点难以检测到。此外,弯枝藻属仅含在全球分布的单一物种,其分子多样性水平较低。据李强等的研究,淡水红藻物种爆发的时间大约在450~600 Mya间,大部分淡水红藻类群均在这段时期内形成[31]。因此可推测弯枝藻属的基因已在早期发生进化后被固定,因此现在未能检测到正选择位点。

表3 各模型参数估计值和对数似然值
当一个基因经受正选择时,表明该类群需要产生新的功能来应对环境的巨变,而当基因处于强烈负选择时,则说明该基因保持原有的重要功能且趋于稳定[13]。通过对弯枝藻属L基因编码蛋白的生物信息学分析,表明Rubisco大亚基二级结构主要由螺旋和折叠构成,结构稳定且结构域十分保守。以往对Rubisco结构的研究表明,Rubisco大亚基的羧基端均有1个由8个螺旋和8个折叠组成的/桶结构域,与相邻的氨基端功能结构域(由2个螺旋和5个折叠组成的小亚基)共同构成酶的活性中心[32]。Rubisco大亚基在植物光合作用中起十分重要的作用,这些结构保证了Rubisco大亚基的重要功能位点保持稳定状态。
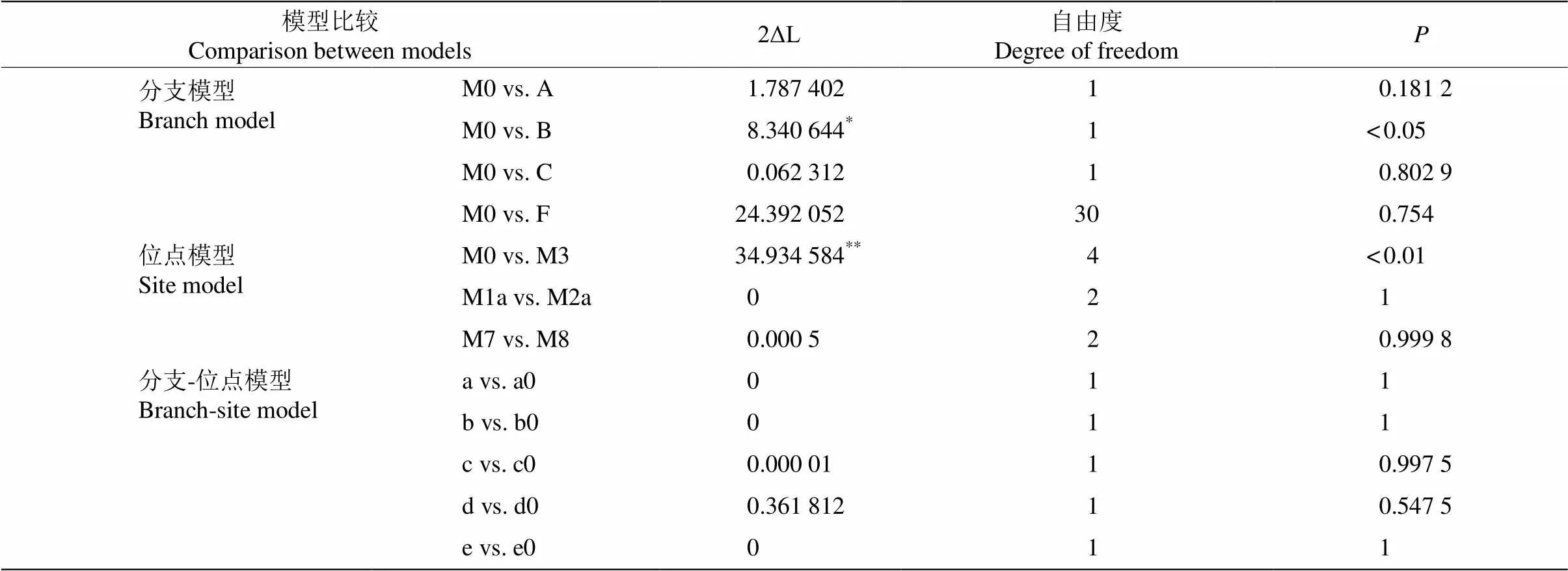
表4 LRT检验统计
*:<0.05; **:<0.01.
在本研究中,位点模型和分支-位点模型中均检测出可能的正选择位点,但经过似然比检验,备择假设不可靠,拒绝存在正选择位点的假设。在过去的几十年里对基因进化中正选择位点的研究成为热点,其中的主要原因是在各模型中正选择/负选择位点判断的统计方法有了很大进步[11,28]。但据报 道[33–34],似然比检验的结果依赖于模型使用的初始参数值,有时很难得到给定模型参数的最大似然估计值,得到的结果可能出现假阳性。Zhang[35]就通过计算机模拟的方法检验出分支-位点模型的似然比检验可能存在假阳性。这种不可靠性可能是由于它对实验中所作假设的违背过于敏感,例如在不同位点分布有不同选择压力,以及在同义替换和非同义替换中的转换、颠换率存在差异所造成的。之后,又对分支-位点模型进行了改进,并使用它构建了两个LRT检验,分别为test1和test2,经验证test2应用于实际数据分析较可靠,很好地解决了假阳性问题[30]。基于已有研究,我们推测可能由于系统发育树分支长度较短,序列数量不够庞大导致假阳性存在,此外,选择压力放松也有可能造成这一结果。
分支模型中各分支的值均小于1,说明整体的弯枝藻属类群处于较强的负选择压力下,但分支A、B、C的进化速率存在一定差异,分支C的值极低(0.000 10),甚至低于单比率模型值(0.027 85), 而分支B的进化速率(0.571 50)则远高于其他分支, LRT检验也证实此观点可靠。这可能是弯枝藻作为全球范围分布的单一物种,从系统发育树分支可以看出其分布具有明显的地域特点,A分支分布于中国、澳大利亚及巴西,B分支分布于美国,C分支分布于马来西亚、印尼、西太平洋岛群等。由于弯枝藻生存的环境通常较为封闭,水体的温度、清洁度、溶氧量以及其他因素均有较大区别,所以长期处于不同的水体环境中可能导致不同分支的进化速率产生差异。
本研究对弯枝藻属L基因的研究支持其未发生适应性进化的观点。在高等植物中,L基因正选择位点的存在十分普遍[12–15]。但目前对藻类的适应性进化研究较少。巩超彦等在淡水红藻串珠藻目(Batrachospermales)植物适应性进化研究中,检测到3个正选择位点,其余位点则普遍处于负选择压力下[10]。因此,今后有必要进一步对淡水红藻其它类群叶绿体L基因的适应性进化进行深入研究,以探究其在海陆变迁过程中如何适应环境的巨变。
[1] SKUJA H. Comments on fresh-water rhodophyceae [J]. Bot Rev, 1938, 4(12): 665–676. doi: 10.1007/BF02869845.
[2] KUMANO S.Freshwater red Algae of the World [M]. Bristol, England: Biopress Limited, 2002: 223–228.
[3] SHI Z X. Flora Slgarum Sinicarum Aquae Dulcis, Vol. 13, Rhodophyta Phaeophyta [M]. Beijing: Science Press, 2006: 1–34. 施之新. 中国淡水藻志, 第13卷, 红藻门, 褐藻门[M]. 北京: 科学出版社, 2006: 1–34.
[4] GAO Y F, NAN F R, FENG J, et al. Review of systematics of Compso- pogonales (Rhodophyta) [J]. World Sci-Technol Res Devel, 2015, 37(3): 314–318. doi: 10.16507/j.issn.1006-6055.2015.03.021.高一帆, 南芳茹, 冯佳, 等. 弯枝藻目Compsopogonales系统分类研究进展[J]. 世界科技研究与发展, 2015, 37(3): 314–318. doi: 10. 16507/j.issn.1006-6055.2015.03.021.
[5] NECCHI O Jr, FO A S G, SALOMAKI E D, et al. Global sampling reveals low genetic diversity within(Compsopogonales, Rhodophyta) [J]. Eur J Phycol, 2013, 48(2): 152–162. doi: 10.1080/ 09670262.2013.783626.
[6] KAPRALOV M V, FILATOV D A. Widespread positive selection in the photosynthetic Rubisco enzyme [J]. BMC Evol Biol, 2007, 7: 73. doi: 10.1186/1471-2148-7-73.
[7] SAGE R F, SEEMANN J R. Regulation of ribulose-1,5-bisphosphate carboxylase/oxygenase activity in response to reduced light intensity in C4plants [J]. Plant Physiol, 1993, 102(1): 21–28. doi: 10.1104/pp.102. 1.21.
[8] SIQUEIRA A S, LIMA A R J, DALL’AGNOL L T, et al. Comparative modeling and molecular dynamics suggest high carboxylase activity of thesp. CACIAM14L protein [J]. J Mol Model, 2016, 22: 68. doi: 10.1007/s00894-016-2943-y.
[9] HARTMAN F C, HARPEL M R. Structure, function, regulation, and assembly of d-ribulose-1,5-bisphosphate carboxylase/oxygenase [J]. Annu Rev Biochem, 1994, 63: 197–234. doi: 10.1146/annurev.bi.63. 070194.001213.
[10] GONG C Y, NAN F R, FENG J, et al. Adaptive evolutionary analysis onL gene of Batrachospermales [J]. Oceanol Limnol Sin, 2017, 48 (3): 527–535. doi: 10.11693/hyhz20161200277.巩超彦, 南芳茹, 冯佳, 等. 串珠藻目植物L基因的适应性进化分析[J]. 海洋与湖沼, 2017, 48(3): 527–535. doi: 10.11693/hyhz201 61200277.
[11] NEI M, KUMAR S. Molecular Evolution and Phylogenetics [M]. New York: Oxford University Press, 2000: 238–273.
[12] ZHOU Y, WANG B, GAO L, et al. Adaptive evolution and coevolution of theL gene in xeric Pteridaceae ferns [J]. Plant Sci J, 2011, 29(4): 409–416. doi: 10.3724/SP.J.1142.2011.40409.周媛, 王博, 高磊, 等. 凤尾蕨科旱生蕨类L基因的适应性进化和共进化分析[J]. 植物科学学报, 2011, 29(4): 409–416. doi: 10. 3724/SP.J.1142.2011.40409.
[13] ZHANG L J, CHEN J, WANG T. Adaptive evolution in the chloroplast gene4 in ferns [J]. Bull Bot Res, 2010, 30(1): 42–50. 张丽君, 陈洁, 王艇. 蕨类植物叶绿体4基因的适应性进化分析[J]. 植物研究, 2010, 30(1): 42–50.
[14] XIONG Y, ZHAO C Y, YANG Q S, et al. In silico cloning, bioinfor- matics and adaptive evolution analysis ofL gene in[J]. Biotechnology, 2014, 24(6): 50–56. doi: 10.3969/j.issn. 1004-311X.2014.06.0137-7.熊勇, 赵春艳, 杨青松, 等. 黄花蒿L基因电子克隆、生物信息学及适应性进化分析[J]. 生物技术, 2014, 24(6): 50–56. doi: 10. 3969/j.issn.1004-311X.2014.06.0137-7.
[15] LIU N, WANG Q B, CHEN J, et al. Adaptive evolution and structure modeling ofL gene in[J]. Chin Sci Bull, 2010, 55(22): 2341–2346. doi: 10.1007/s11434-010-3023-9.刘念, 王庆彪, 陈婕, 等. 麻黄属L基因的适应性进化检测与结构模建[J]. 科学通报, 2010, 55(14): 1341–1346. doi: 10.1007/s 11434-010-3023-9.
[16] JIANG B, GAO L, LI J, et al. Adaptive evolution of the chloroplast genome in AA-genomespecies [J]. Chin Sci Bull, 2014, 59(20): 1975–1983. doi: 10.1360/N972014-00127.姜斌, 高磊, 李佳, 等. 稻属AA型物种叶绿体基因组的适应性进化[J]. 科学通报, 2014, 59(20): 1975–1983. doi: 10.1360/N972014- 00127.
[17] THOMPSON J D, GIBSON T J, PLEWNIAK F, et al. The CLUSTAL_X windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools [J]. Nucl Acids Res, 1997, 25(24): 4876–4882. doi: 10.1093/nar/25.24.4876.
[18] KUMAR S, STECHER G, TAMURA K. MEGA7: Molecular evolu- tionary genetics analysis version 7.0 for bigger datasets [J]. Mol Biol Evol, 2016, 33(7): 1870–1874. doi: 10.1093/molbev/msw054.
[19] POSADA D, CRANDALL K A. MODELTEST: Testing the model of DNA substitution [J]. Bioinformatics, 1998, 14(9): 817–818. doi: 10. 1093/bioinformatics/14.9.817.
[20] GUINDON S, DUFAYARD J F, LEFORT V, et al. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0 [J]. Syst Biol, 2010, 59(3): 307–321. doi: 10.1093/sysbio/syq010.
[21] RANNALA B, YANG Z. Probability distribution of molecular evolu- tionary trees: A new method of phylogenetic inference [J]. J Mol Evol, 1996, 43(3): 304–311. doi: 10.1007/BF02338839.
[22] BURLAND T G. DNASTAR’s Lasergene sequence analysis software [M]// MISENER S, KRAWETZ S A. Bioinformatics Methods and Protocols: Methods in Molecular Biology™, Vol. 132. Totowa, NJ: Humana Press, 2000: 71–91. doi: 10.1385/1-59259-192-2:71.
[23] XIONG Y, ZHAO C Y, YANG Q S, et al. Molecular cloning, bioinfor- matics analysis ofL gene in[J]. N Hort, 2015, 39(4): 89–95. doi: 10.11937/bfyy.201504021.熊勇, 赵春艳, 杨青松, 等. 雏菊L基因的克隆及其生物信息学分析[J]. 北方园艺, 2015, 39(4): 89–95. doi: 10.11937/bfyy.201504 021.
[24] YANG Z. Computational Molecular Evolution [M]. Oxford: Oxford University Press, 2006: 56–167.
[25] YANG Z H, SWANSON W J, VACQUIER V D. Maximum-likelihood analysis of molecular adaptation in abalone sperm lysin reveals variable selective pressures among lineages and sites [J]. Mol Biol Evol, 2000, 17(10): 1446–1455. doi: 10.1093/oxfordjournals.molbev. a026245.
[26] WONG W S, YANG Z H, GOLDMAN N, et al. Accuracy and power of statistical methods for detecting adaptive evolution in protein coding sequences and for identifying positively selected sites [J]. Genetics, 2004, 168(2): 1041–1051. doi: 10.1534/genetics.104.031153.
[27] YANG Z. PAML: A program package for phylogenetic analysis by maximum likelihood [J]. Comput Appl Biosci, 1997, 13(5): 555–556.
[28] YANG Z. Likelihood ratio tests for detecting positive selection and application to primate lysozyme evolution [J]. Mol Biol Evol, 1998, 15(5): 568–573. doi: 10.1093/oxfordjournals.molbev.a025957.
[29] YANG Z H, BIELAWSKI J P. Statistical methods for detecting mole- cular adaptation [J]. Trends Ecol Evol, 2000, 15(12): 496–503. doi: 10. 1016/S0169-5347(00)01994-7.
[30] ZHANG J Z, NIELSEN R, YANG Z H. Evaluation of an improved branch-site likelihood method for detecting positive selection at the molecular level [J]. Mol Biol Evol, 2005, 22(12): 2472–2479. doi: 10. 1093/molbev/msi237.
[31] LI Q, JI L, XIE S L. Phylogenetic analysis of Batrachospemales (Florideophyceae, Rhodophyta) based on chloroplastL sequences [J]. Acta Hydrobiol Sin, 2010, 34(1): 20–28. doi: 10.3724/SP.J.1035. 2010.00020.李强, 吉莉, 谢树莲. 串珠藻目植物的系统发育: 基于L序列的证据[J]. 水生生物学报, 2010, 34(1): 20–28. doi: 10.3724/SP.J.1035. 2010.00020.
[32] KNIGHT S, ANDERSSON I, BRÄNDÉN C I. Crystallographic analysis of ribulose 1,5-bisphosphate carboxylase from spinach at 2·4 Å resolution: Subunit interactions and active site [J]. J Mol Biol, 1990, 215(1): 113–160. doi: 10.1016/S0022-2836(05)80100-7.
[33] SUZUKI Y, NEI M. Reliabilities of parsimony-based and likelihood- based methods for detecting positive selection at single amino acid sites [J]. Mol Biol Evol, 2001, 18(12): 2179–2185. doi: 10.1093/ oxfordjournals.molbev.a003764.
[34] SUZUKI Y, NEI M. Simulation study of the reliability and robustness of the statistical methods for detecting positive selection at single amino acid sites [J]. Mol Biol Evol, 2002, 19(11): 1865–1869. doi: 10. 1093/oxfordjournals.molbev.a004010.
[35] ZHANG J. Frequent false detection of positive selection by the likelihood method with branch-site models [J]. Mol Biol Evol, 2004, 21(7): 1332–1339. doi: 10.1093/molbev/msh117.
Adaptive Evolutionary Analysis of theL Gene from(Rhodophyta)
HAN Yu-xin, NAN Fang-ru, GONG Chao-yan, FENG Jia, LÜ Jun-ping, LIU Qi, XIE Shu-lian*
(School of Life Science, Shanxi University, Taiyuan 030006, China)
In order to reveal the chloroplast gene and adaptive evolution characters of Rhodophyta, the 17L genes ofand the similar group of freshwater red algae were selected, the bioinformatics ofproteins encoded byL genes ofwere analyzed by using software PAML4.9, and the selection sites ofgenes were detected by using branch model, site model and branch-site models. The results showed that the secondary structure of protein encoded byL ofwas mainly composed of α helix and β folding, so its structure was very stable.The phylogenetic tree with the maximum likelihood method showed that the inner group had only one species, could be divided into three small branches, and they had obvious geographical distribution. No significant positively selected sites were detected under all three evolutionary models, indicating that most of the sites were under negative selection pressure. Therefore, there is no adaptive evolution ofL gene in.
;L gene; Protein structure prediction; Adaptive evolution
10.11926/jtsb.3909
2018-03-19
2018-04-29
国家自然科学基金项目(31670208, 31370239)资助
This work was supported by the National Natural Science Foundation of China (Grant No. 31670208, 31370239).
韩雨昕(1993~ ),女,硕士研究生,主要从事藻类学研究。E-mail: 15835107990@163.com
E-mail: xiesl@sxu.edu.cn
