Comprehensive multi-omics analysis identified core molecular processes in esophageal cancer and revealed GNGT2 as a potential prognostic marker
2019-02-12GuoMinLiuXuanJiTianChengLuLiWeiDuanWenYuanJiaYunLiuMaoLeiSunYunGangLuo
Guo-Min Liu, Xuan Ji, Tian-Cheng Lu, Li-Wei Duan, Wen-Yuan Jia, Yun Liu, Mao-Lei Sun, Yun-Gang Luo
Abstract
Key words: Esophageal cancer; Molecular pathogenesis; Enrichment analysis; Gene interaction module; Regulatory factors; GNGT2
INTRODUCTION
Esophageal cancer is one of the world's most common cancers with poor diagnosis and high mortality because of invasiveness and a fast growth rate[1]. From a therapeutic point of view, esophageal cancer can be divided into early esophageal cancer, locally advanced resectable esophageal cancer, locally advanced unresectable esophageal cancer, and metastatic esophageal cancer. Because of the anatomical features of esophageal cancer, esophageal cancer is usually detected in the late stage,which vitally affects the treatment options and prognosis of patients[2].
During the development of esophageal cancer, the rs11473 polymorphism of the miR-483-5p binding site plays a vital role in the 3'-untranslated region of the basigin gene[3]. Single nucleotide polymorphisms in telomerase reverse transcriptase may be associated with susceptibility to esophageal cancer and contribute to the development of esophageal cancer[4]. MiR-20b may play an essential role in the tumorigenesis of esophageal cancer by regulating phosphatase and tensin homolog expression, which may be a potential therapeutic target for the treatment of esophageal cancer[5].Growing evidence has revealed molecular targets for diagnosis and prognosis using bioinformatic analysis in the field of oncology[6-16]. These findings have deepened our understanding of the pathogenesis of esophageal cancer and have guided the direction of our research. However, the molecular pathogenesis of the disease is still elusive.
To explore comprehensively the molecular processes and potential therapeutic targets of esophageal cancer progression, we conducted a systematic module analysis.Overall, our work details the role of multifactorial mediated dysfunction modules in the growth of esophageal cancer and identifies essential genes and related biological processes, finding potential molecular mechanisms and therapeutic targets [G protein subunit gamma transducin 2 (GNGT2)] for esophageal cancer.
MATERIALS AND METHODS
Patient samples and cell lines
All esophageal cancer analyses in this study involving human participants were in accordance with the ethical standards of the Second Hospital of Jilin University and with the Declaration of Helsinki. A total of 40 esophageal cancer patients and healthy control volunteers, who matched for age and sex, were involved in this study.Informed consent was obtained from all participants. The esophageal cancer cell lines EC109 and KYSE70 were kindly provided by Laboratory in The Second Hospital of Jilin University. The cells were maintained in RPMI-1640 medium containing 10%fetal bovine serum.
Quantitative real-time PCR and cell proliferation experiment
Total RNA was extracted from case/control group using TRIzol. The quantitative real time-polymerase chain reaction (PCR) experiment was conducted in a real-time PCR detection system using SYBR Green qPCR Master Mix. Primers were designed and synthesized by Novogene (Beijing, China). Glyceraldehyde 3-phosphate dehydrogenase was used as an internal control. Cell counting kit-8 assay was used to measure cell proliferation. Transfected cells were cultured for 0-96 h and incubated at 37 °C for 2 h. A spectrophotometer (450 nm) was used to quantitate samples.
Data resource
The Cancer Genome Atlas (TCGA) is a joint project of the National Cancer Institute and the American Human Genome Research Institute. High-throughput genomic analysis technology is a useful tool for people to understand better cancer, and it improves their abilities to prevent, diagnose, and treat disease. We downloaded esophageal cancer RNA-Seq data from the TCGA database and screened non-coding RNA (ncRNA)-mRNA interaction pairs with a score ≥ 0.5 from RNA Associated Interaction Database v2.0[17], including 431937 interacting pairs involving 5431 ncRNAs. All human transcription factor target data were downloaded and used in the general database-Transcriptional Regulatory Relationships Unraveled by Sentencebased Text mining v2 database for transcriptional studies, including 2492 transcription factors and 9396 interaction pairs.
Differential expression analysis
In order to explore the molecular process of esophageal cancer staging, we selected four stages of esophageal cancer and normal samples for differential expression analysis, including healthy tissue samplesvsstage 1 disease samples, stage 1 disease samplesvsstage 2 disease samples, stage 2 disease samplesvsstage 3 disease samples,and stage 3 disease samplesvsstage 4 disease samples. We used the limma package for analysis[18-20]. Using the Correct background function, we performed background correction and normalization on the data. The normalize Between Arrays function quantile normalization method can filter out the control probe and the low expression probe. The differentially expressed genes of the data set were identified based on the lmFit and eBayes functions (P< 0.01) using default parameters.
Establishing a protein interaction network to identify esophageal cancer related functional modules
A protein-protein interaction system was constructed based on Search Tool for the Retrieval of Interacting Genes/Proteins database data (score > 500). The gene module of more than 30 nodes was screened throughout the network using the ClusterONE plug-in[18]of the Cytoscape software[19]. We use the Cytoscape plugin CytoHubba[20]to identify hub genes in the module subnet, while CytoHubba contains 12 methods for identifying hub genes. We obtained the top 10 genes and then screened the repeat genes in the 12 sets of genes for survival analysis.
Enrichment analysis
The study of the functions and signal transduction pathways involved in genes contributes to our understanding of the molecular mechanisms of disease. Gene ontology function and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis was performed using the R language Cluster profiler package[21].The Cluster profiler is a bioconductor software package that provides statistical analysis of functional clustering of gene sets.
Predictive transcriptional factors and ncRNAs for significant regulatory modules
The transcription and post-transcriptional regulation of genes are often dominated by the regulation of transcription factors (TF) and ncRNA. Therefore, we have scientifically predicted its role in the esophageal cancer dysfunction module. If the regulatory effect between the regulator and the module exceeds 2, and the number of organizational relationships between the regulator and the module is essential(hypergeometric test,Pvalue < 0.01), it can be confirmed that it is a regulator of the critical regulatory module.
RESULTS
Identifying the expression of dysregulated molecules in esophageal cancer
Biologists have conducted many experiments and studies on the pathogenesis of esophageal cancer and have thus identified potential pathogenic genes for the deterioration of esophageal cancer. To observe molecular changes in the progression of esophageal cancer, we performed differential expression analysis based on RNASeq data from four stages of esophageal cancer in the TCGA database. Based on analysis of phase 1 disease samples of normal tissue samples and esophageal cancer,analysis of phase 1 disease samples and stage 2 disease samples, analysis of stage 2 disease samples and phase 3 disease samples, and analysis of stage 3 disease samples and stage 4 disease samples, we obtained differential expression genes (DEGs)associated with each stage of esophageal cancer. A total of 7457 differentially expressed genes were received (Figure 1B). We believe that the presence of these differentially expressed genes is closely related to the development of various stages of esophageal cancer. Of the 7457 DEGs, there were 13 common genes (Table 1). The genes that were continuously down-regulated areCPLX2, DPEP1, EPHA5, SCGB1A1,ST18. The genes that were continuously up-regulated areFGF14, KCNH6,LOC100506136, RGS7, SH3GL2, THBS4(Figure 1A).
Identify functional esophageal cancer staging related modules
Gene module analysis helps us to study the complex collaborative relationships between multiple genes. Based on the protein interaction data of the STING database,the interaction network of differentially expressed genes was constructed, and 14 functional barrier modules were explored. Using the 12 methods in Cyto-hubba, a total of 758 hub genes were identified in the interaction sub-network of the module genes, including the geneSH3GL2, which is continuously up-regulated in Module 8.Further, 23 hub genes shared by the top10 gene set in 12 methods were screened for survival analysis. The results show thatGNGT2in module 6 is the related gene (P=0.014) (Figure 2A). A decrease in survival rate accompanied the high expression ofGNGT2gene, and the expression level ofGNGT2gene was negatively correlated with survival rate. Function and pathway are essential mediators of the physiological response of the disease. We performed GC enrichment analysis on 14 module genes(Figure 2B) and KEGG (Figure 2C). The main biological processes include positive regulation of protein transport, gastric acid secretion, and insulin-like growth factor receptor binding. The main signal transduction pathways involved are the p53 signal transduction, the epidermal growth factor signal transduction, and the epidermal growth factor receptor signal transduction pathways. These pathways play crucial roles in the dysfunctional module for the functions and pathways involved in multiple genes.
TFs and ncRNAs that drive esophageal cancer progression
From the perspective of systems biology and systems genetics, transcription and posttranscriptional regulation of genes have long been recognized as crucial regulators of disease development, while transcription factors and ncRNAs are universal regulators of expression and function. Although the role of TFs and ncRNA regulation of esophageal cancer progression has been evaluated by many biologists, few studies have focused on their overall global effect on dysfunctional mechanisms and the role they play in development. Therefore, in this study, based on the targeted regulationrelationship between TF and ncRNA on the module gene, we performed a pivotal analysis of the conventional module to explore the crucial regulator that regulates the progression of esophageal cancer. The results showed that a total of 54 transcription factors involved 54 TF-module target pairs and 853 ncRNAs involved 944 ncRNA-module regulatory pairs. Statistical analysis revealed that TF HIF1A and ncRNA CRNDE regulate the most dysfunctional modules. These crucial transcription factors and ncRNAs may influence the development and progression of esophageal cancer by mediating dysfunctional modules. Thus, we identified these potential factors as regulators of dysfunction in esophageal cancer. Notably, hsa-miR-330-3p up-regulates the differentially expressed geneSH3GL2throughout the esophageal cancer process,suggesting that hsa-miR-330-3p plays a crucial role in four stages of esophageal cancer.
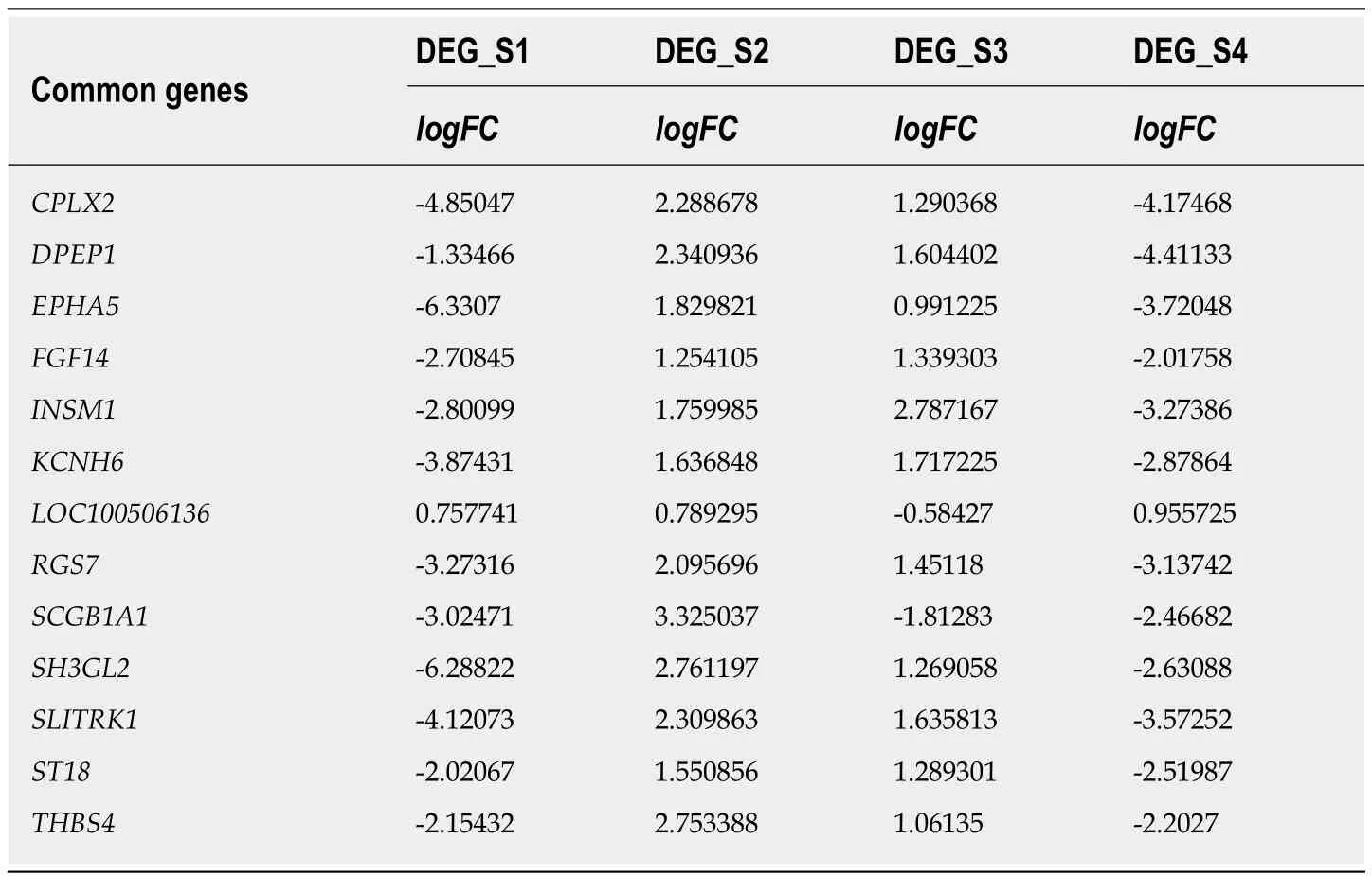
Table 1 Differential expression of 13 common genes in four different stages of esophageal cancer
GNGT2 expression is upregulated in the esophageal epithelial cells of esophageal cancer and cell lines
To investigate changes inGNGT2expression during esophageal cancer development,samples from esophageal cancer patients (n= 20) and esophagus controls (n= 20)were subjected to quantitative real time-PCR analysis. As shown in Figure 3A,expression ofGNGT2gene was significantly upregulated in the esophageal epithelial cells of esophageal cancer patients. The experiment in esophageal cancer cell lines(EC109 and KYSE70) showed consistent results (Figure 3B) (P< 0.05).
GNGT2 could promote the proliferation of esophageal cancer cell lines
To explore further the role of GNGT2 in the proliferation of EC109 and KYSE70 cells,cells were transfected withGNGT2siRNA. As shown in Figure 3C,GNGT2mRNA expression level was significantly decreased in EC109 and KYSE70 cells. Moreover,the proliferation ofGNGT2transfected group was significantly lower than that of the control group (Figure 3D). Taken together, the above results demonstrated that GNGT2 could promote the proliferation of esophageal cancer cell lines.
DISCUSSION

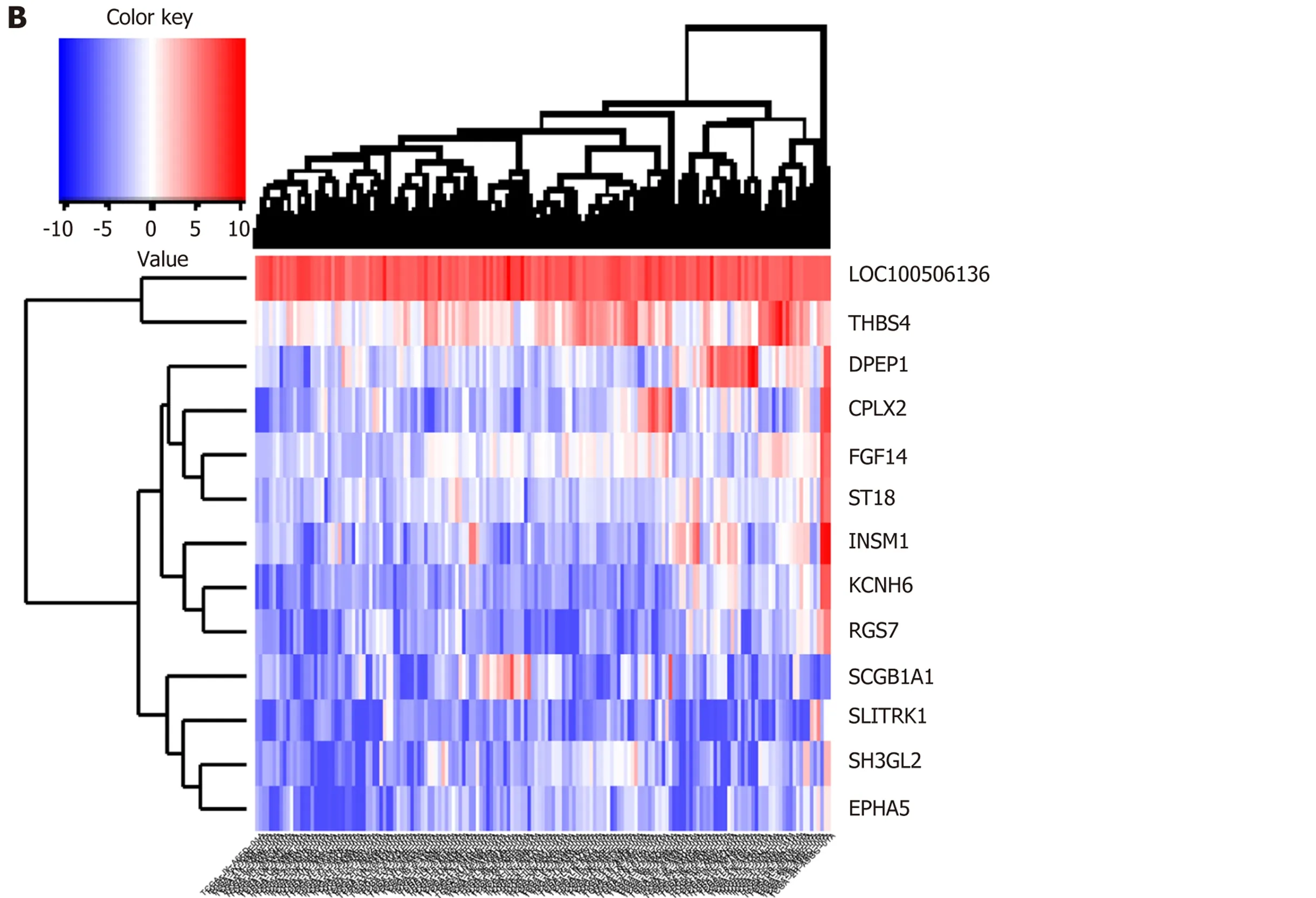
Figure 1 Synergistic expression of differential genes in four samples of esophageal cancer in patient samples. A: Continuous regulation of common genes in differentially expressed genes in four stages; B: Expression heat map of common genes in samples of differentially expressed genes in four stages.
Esophageal cancer is one of the most deadly cancers, mainly because it is extremely aggressive and has a poor survival rate. Its 5-year survival rate is about 15%-25%[1].The underlying cause of this disappointing low survival rate is that most patients have reached the late stage of disease at the time of detection. For patients with metastatic and unresectable disease, their chances of survival are limited[22]. In the present study, we collected RNA-Seq data from TCGA esophageal cancer and selected four stages of esophageal cancer disease samples and normal samples for differential analysis, and obtained four sets of time series differentially expressed genes. After screening, we found 13 common genes in four groups of DEGs. Komatsuet al[23]studied clinical biomarkers of pulmonary neuroendocrine tumors (LNET) and found that CPLX2 was strongly positive in 16.3% of the examination groups. Importantly,positive CPLX2 expression is associated with lymphatic invasion, pathological staging, and adverse disease-specific survival in LNET patients. It was concluded that CPLX2 is a novel clinical biomarker for LNET[23]. In the study of breast cancer diagnostic markers, Fuet al[24]found that changes in gene expression, such as DPEP1,may lead to cancer progression. DPEP1 has been identified as a prognostic gene for colorectal cancer (CRC). We found thatDPEP1is overexpressed in CRC. After knocking out theDPEP1gene, cells (SW480 and HCT116) exhibited increased apoptosis and attenuated cell proliferation and cell invasion[25]. In the study of CRC,Eisenach[26]found that the expression of DPEP1 was increased in CRC tissues compared with normal mucosa. Zhanget al[27]also noted the DPEP1 gene in the study of pancreatic ductal adenocarcinoma and found that its gene expression was negatively correlated with histological grade and that lower expression ofDPEP1in tumors was associated with poor survival. Chenet al[28]analyzed the gastric cancerassociated Gene Expression Omnibus data and found that thrombospondin 4 (THBS4)was up-regulated in patients with recurrent gastric cancer and was positively correlated with the pathological stage and poor prognosis of gastric cancer. THBS4 stimulates the proliferation of gastric cancer cells. The breast-related gene explored by Huanget al[29]contains the geneTHBS4, which is up-regulated in breast cancer. In the study of hepatocellular carcinoma, Suet al[30]found that knockdown of THBS4 inhibited migration and invasion of hepatocellular carcinoma cells as well as hemangiocarcinoma-induced angiogenesis. THBS4 as a target is very promising for the treatment of advanced liver cancer. Both of the above genes were present in the differential genes of the four stages of esophageal cancer in this study and were continuously down-regulated. Moreover, THBS4 was identified as a clinical biomarker gene and a therapeutic target gene in various cancers. Therefore, we can reasonably speculate that this gene plays an important role in the occurrence and development of esophageal cancer, providing a reasonable direction for further study of esophageal cancer.
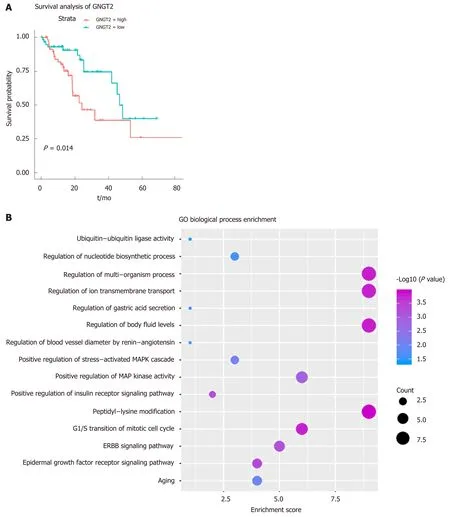
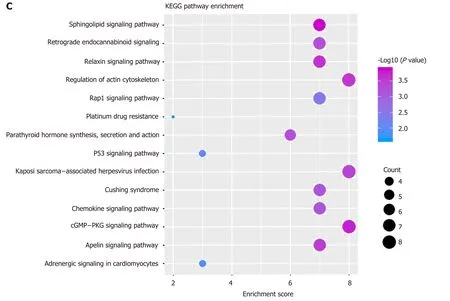
Figure 2 Dysfunctional modules. A: Survival analysis of G protein subunit gamma transducin 2; B and C: Module gene function and pathway enrichment analysis.The larger the circle, the greater the proportion of the gene in the Gene Ontology/Kyoto Encyclopedia of Genes and Genomes. GNGT2: G protein subunit gamma transducin 2; MAPK: Mitogen-activated protein kinase; ERBB: Epidermal growth factor.
The results of the methylation test showed that theSSTgene was up-regulated extensively, which may be a key gene involved in methylation modification to regulate the progression of esophageal cancer. Jinet al[31]found that hypermethylation of theSSTpromoter is common and is associated with early tumor progression in Barrett's esophagus. TheSH3GL2gene is up-regulated. The gene is not only the common DEGs of the four-stage time series but also the Hub gene in module 6. It also may play an important role in the regulation of esophageal cancer by methylation modification. Ghoshet al[32]studied the effect of SH3GL2 methylation on the pathogenesis of head and neck squamous cell carcinoma, and abnormalSH3GL2is an independent pathway for early developmental abnormalities of the head and neck.
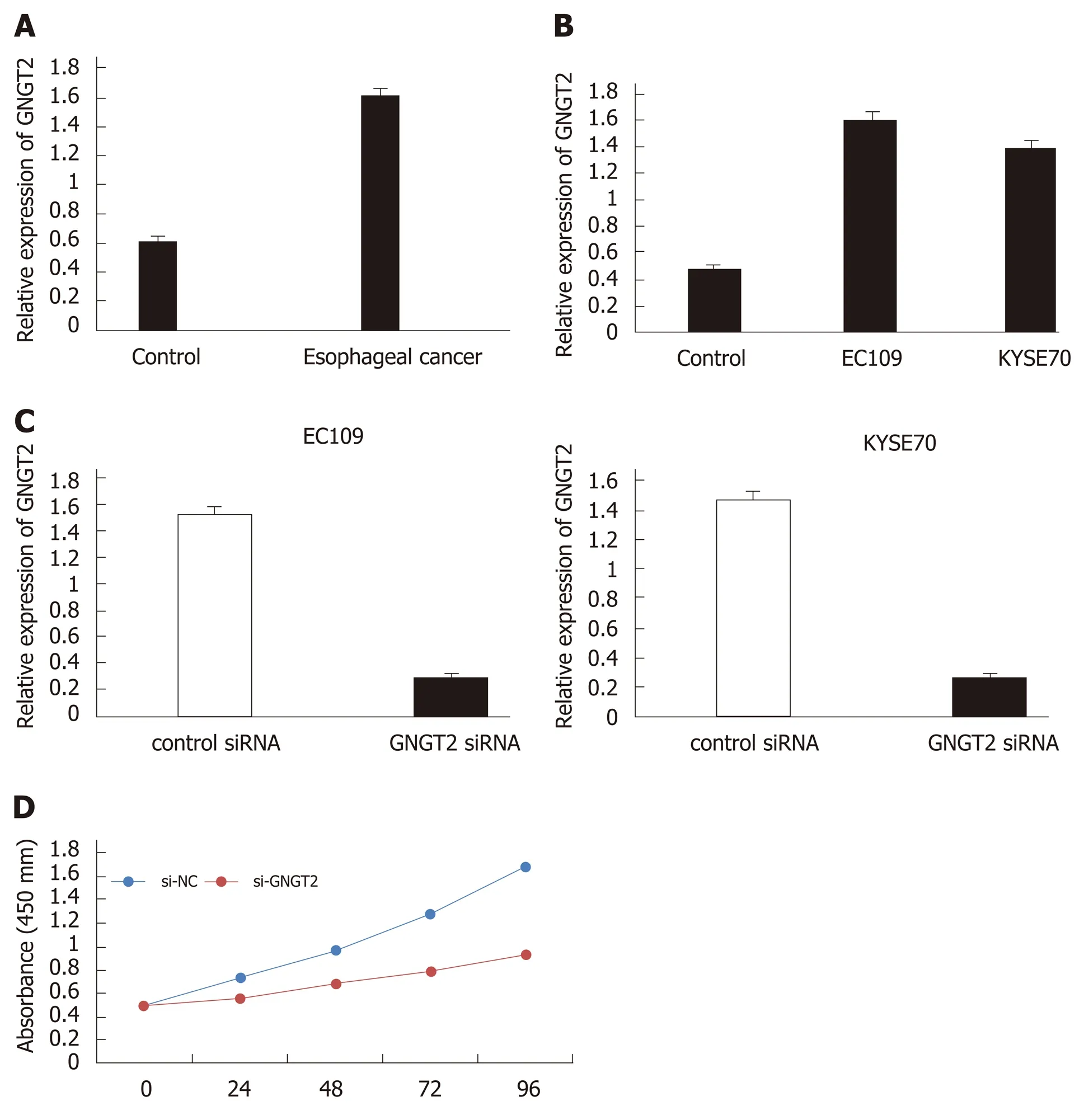
Figure 3 Molecular mechanism and expression of G protein subunit gamma transducin 2 in esophageal cancer. A and B: The expression of G protein subunit gamma transducin 2 (GNGT2) in esophageal cancer patients and cell lines; C: Transfected of EC109 and KYSE70 cells; D: GNGT2 promote the proliferation of esophageal cancer cells.
ARTICLE HIGHLIGHTS

Overall, our work describes in detail the role of multifactor-mediated dysfunction module in the whole process of esophageal cancer, identifying key genes for staging and related biological processes, which may help to identify potential molecular mechanisms and therapeutic targets for the deterioration of esophageal cancer.
Research perspectives
This work not only helps us to reveal the potential regulatory factors involved in the development of disease but also deepen our understanding of its deterioration mechanism.
杂志排行

World Journal of Gastroenterology的其它文章
- Gastric electrical stimulation: An emerging therapy for children with intractable gastroparesis
- Diagnostic and prognostic value of lncRNA cancer susceptibility candidate 9 in hepatocellular carcinoma
- Operative complications and economic outcomes of cholecystectomy for acute cholecystitis
- Hepatitis C virus eradication with directly acting antivirals improves health-related quality of life and psychological symptoms
- Significance of postoperative follow-up of patients with metastatic colorectal cancer using circulating tumor DNA
- Pulmonary tumor thrombotic microangiopathy of hepatocellular carcinoma: A case report and review of literature