超高效液相色谱法同时检测茶叶中的没食子酸、咖啡碱和儿茶素
2019-02-06赵明明周聪彭茂民周有祥
赵明明 周聪 彭茂民 周有祥


摘要:通过优化前处理提取条件和超高效液相色谱分离方法,建立了茶叶中没食子酸、咖啡碱和8种儿茶素定性和定量方法。结果表明,选用ACQUITY UPLC BEH C18(1.7 μm,2.1 mm×50 mm)色谱柱,色谱条件为柱温30 ℃,流速0.2 mL/min,流动相选择0.1%甲酸水-0.1 %甲酸甲醇溶液,检测波长278 nm,10种组分在0.05~10.00 mg/L具有良好的线性关系,绿茶样品中各组分含量的相对偏差RSD在1.77%~5.89%,表明该方法简洁快速,且精密度好。
关键词:茶叶;超高效液相色谱法;没食子酸;咖啡碱;儿茶素
中图分类号:S571.1;O657.7+2 文献标识码:A
文章编号:0439-8114(2019)24-0205-04
DOI:10.14088/j.cnki.issn0439-8114.2019.24.050 开放科学(资源服务)标识码(OSID):
Simultaneous determination of gallic acid,caffeine and catechins in tea by UPLC
ZHAO Ming-ming,ZHOU Cong,PENG Mao-min,ZHOU You-xiang
(Institute of Quality Standard and Testing Technology for Agro-Products,Hubei Academy of Agricultural Sciences,Wuhan 430064,China)
Abstract: A qualitative and quantitative method for gallic acid, CAF and 8 catechins in tea were established by optimizing the extraction conditions of pretreatment and separation methods of Ultra Performance Liquid Chromatography(UPLC). The results showed that the ACQUITY UPLC BEH C18(1.7 μm,2.1 mm×50 mm) chromatographic column was used and the chromatographic conditions were as follows:The column temperature was 30 ℃, the flow rate was 0.2 mL/min, the mobile phase was 0.1% formic acid water -0.1% formic acid methanol solution, the detection wavelength was 278 nm. The 10 components in the range of 0.05~10.00 mg/L had a good linear relationship, the relative deviation of the content of each component in green tea samples RSD between 1.77%~5.89%. The method is simple, fast and accurate.
Key words: tea; ultra high performance liquid chromatography; catechins; caffeine; gallic acid
茶叶中所含的多酚类物质是一类存在于茶水中的多元酚的混合物,其中最重要的是以儿茶素为主体的黄烷醇类,其含量占多酚类总量的70%~80%[1],是茶树次生物质代谢的重要成分,对茶叶的色、香、味品质的形成有重要作用。酚酸是具有羧基和羟基的芳香族化合物,早在19世纪中叶就开始了茶叶中酚酸的研究,1847年从绿茶中分离出没食子酸[2],没食子酸在茶叶中的含量为干物质的0.5%~1.4%[3]。生物碱是茶叶重要的代谢产物,主要包括咖啡碱、可可碱和茶碱,在茶叶干物质中含量最多[4]。这些营养物质对人体具有极好的功效价值,如儿茶素抗氧化、保护心脑血管、抗肿瘤等作用;咖啡碱能兴奋提神、舒张血管等;没食子酸具有强抗氧化性,也有抗肿瘤、抗血栓等药理作用。目前,关于茶叶没食子酸、儿茶素及咖啡碱测定方法的报道已经很多,主要有薄层色谱法、液相色谱-紫外检测、液相色谱-质谱、毛细管区带电泳法等检测方法[5]。高效液相色谱法应用最多,但是同时检测多种成分的方法耗时较长,超高效液相色谱法高效迅速,尚未广泛应用于茶叶中茶多酚及生物碱的分离检测。因此建立快速、高通量、灵敏度高、特异性强的茶叶中没食子酸、儿茶素和咖啡碱含量的超高效液相色谱法测定方法具有十分重要的意义。
本研究主要采用超高效液相色谱(UPLC)法测定普洱茶中儿茶素(C)、表儿茶素(EC)、表没食子儿茶素(EGC)、表儿茶素没食子酸酯(ECG)、表没食子儿茶素没食子酸酯(EGCG)、没食子儿茶素(GC)、兒茶素没食子酸酯(CG)、没食子儿茶素没食子酸酯(GCG)和没食子酸(GA)9种儿茶素和咖啡碱(CAF)组分的含量,该法快速简便,能够同时测定多种儿茶素。
1 材料与方法
1.1 材料
龙毛尖绿茶、九龙利川红茶、大集白茶购自市场。GA、GC、GCG、CG、C、EGCG、EGC、EC、ECG和CAF标准品购自国家标准物质网。
1.2 仪器与试剂
DZKW-4电热恒温水浴锅(北京中兴伟业仪器有限公司),Waters ACQUITY UPLC-PDA超高效液相色谱仪(美国Waters公司)。
甲醇、乙醇等分析纯试剂购自国药集团化学试剂有限公司,色谱纯试剂购自J.T.Baker公司,EDTA溶液,10 mg/mL;抗坏血酸溶液,10 mg/mL。
稳定溶液:分别将25 mL EDTA溶液、25 mL抗坏血酸溶液、50 mL甲醇溶液加入500 mL容量瓶中,用水定容至刻度,摇匀。
1.3 方法
1.3.1 标准溶液的配置 称取适量儿茶素(GA、GC、GCG、CG、C、EGCG、EGC、EC、ECG)和CAF于25 mL容量瓶中,以稳定溶液定容,得到单标母液,于4 ℃保藏,临用前配置混标,于4 ℃保藏。单标浓度为:100 μg/mL GA、100 μg/mL CAF、800 μg/mL GC、800 μg/mL EGC、600 μg/mL C、200 μg/mL EGCG、400 μg/mL EC、400 μg/mL GCG、400 μg/mL ECG、400 μg/mL CG。
1.3.2 前处理方法的优化 样品制备:茶叶样品充分粉碎、过筛,密封常温保存。试验分3组,A组:70%甲醇(80 ℃预热),茶水比1∶100(0.1 g茶叶粉末加10 mL溶剂),80 ℃浸提10 min,浸提过程摇匀以保证浸提完全。3 500 r/min离心10 min,吸取上清液至50 mL容量瓶中,沉淀再重复提取一次,合并上清液,用稳定溶液定容;B组:水提(80 ℃预热),茶水比1∶100,80 ℃浸提10 min,沉淀再用80 ℃水提取一次,合并上清液,用稳定溶液定容;C组:用70%甲醇(80 ℃预热)提取一次,沉淀再用80 ℃水提取一次,合并上清液,用稳定溶液定容。提取液用0.22 μm微孔滤膜过滤,UPLC进样2 μL。
1.3.3 色谱条件的优化
1)色谱柱的选择。选用美国Waters的 ACQUITY UPLC BEH C18(1.7 μm,2.1 mm×100 mm)、ACQUITY UPLC BEH C18(1.7 μm,2.1 mm×50 mm)和ACQUITY UPLC HSS T3(1.8 μm,1.0 mm×100 mm)3种色谱分离柱进行比较。
2)流动相及其梯度的选择。分别以甲醇和乙腈作为流动相,比较出峰情况。在此基础上,比较不同酸度流动相(0.2%甲酸溶液、0.1%甲酸溶液、0.05%甲酸溶液、0.01%甲酸溶液、0.2%乙酸溶液、0.1%乙酸溶液、0.05%乙酸溶液和0.01%乙酸溶液)出峰情况。
3)柱温和流速的选择。选择不同的温度(25、30、35 ℃)用于色谱柱温度条件优化,选择0.1和0.2 mL/min的流速进行流速条件优化。
4)检测波长的选择。分别选择210 nm和278 nm作为检测波长,观察出峰情况。
1.3.4 标准曲线、线性范围及方法的精确度 用稳定溶液将混标溶液进行梯度稀釋,按其所得峰面积的平均值与对应浓度(μg/L)作标准曲线,计算出回归方程和相关系数。按“1.3.2”方法提取茶叶上清液,每个样品测6组平行,计算各组分RSD。
2 结果与分析
2.1 出峰时间的确定
将所配置的儿茶素和咖啡碱的单标与混标色谱图相比较,确定各成分的出峰时间,如图1所示,具体出峰时间见表1。
2.2 前处理条件的优化
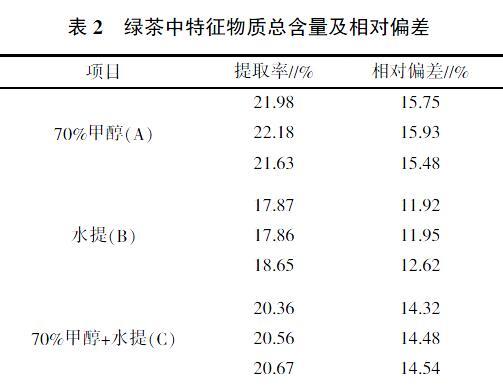
在3组试验组中,采用不同的浸提溶剂组合,结果发现用70%的甲醇提取2次的提取率最高(表2)。这是由于甲醇的羟基与儿茶素及茶多酚分子中含有的酚羟基能形成氢键,具有良好的溶解性[6],而且天然状态的儿茶素与茶多酚往往以氢键与蛋白质、多糖等大分子物质结合成复合物形式存在,甲醇溶液具有断裂氢键的能力,有利于使缔合态的儿茶素及茶多酚游离出来。
2.3 色谱条件的优化
2.3.1 色谱柱的确定 从色谱试验来看,UPLC BEH C18(1.7 μm,2.1 mm×100 mm)的分离效果最好。HSS T3柱是高强度的硅胶,而UPLC BEH C18柱采用桥式亚乙基硅氧烷,亚乙基硅氧烷与极性成分的键合作用较强,有利于没食子酸、儿茶素和咖啡碱的分离,其主要利用分子与层析材料烃基之间的疏水作用来实现分离,其分离效率高。且1.7 μm粒径较1.8 μm粒径小,所以C18柱不论是从峰形方面来说,还是从分离情况来考虑,其效果都更好。而50 mm BEH C18柱所需分离时间比100 mm BEH C18柱所需时间短,效率更高,因而最终选择ACQUITY UPLC BEH C18(1.7 μm,2.1 mm×50 mm)。
2.3.2 流动相及其梯度的确定 乙腈极性较强,对目标物洗脱能力更强,使得大多数目标物很快洗脱,分离度差,短时间内无法实现多种目标物的分离。而使用甲醇有很好的分离度。因此,本方法选用甲醇作为强洗脱相。
由于反相色谱柱填料表面存在残余的羟基,而部分酚类化合物具有弱酸性,能够与残余硅羟基发生静电作用产生次级保留效应,被推迟洗脱下来,产生拖尾现象。酸性环境能够抑制硅羟基电离,向流动相中添加少量酸,能够明显改善峰型[7]。为了获得准确的定量分析结果,试验考察了酸的种类以及酸的浓度对10种目标物质分离效果的影响。结果表明,多酚化合物在0.1%甲酸水和0.1%甲酸甲醇梯度洗脱条件下,都能够出峰且基线平稳(表3)。
2.3.3 柱温和流速的确定 温度是反相色谱中一个控制选择性的重要变量,在高温下,色谱峰的拖尾现象得到改善,峰形变得更加对称,在C18柱上对儿茶素进行分析,无论是以纯水作为流动相,还是以水和有机溶剂共同作为流动相,色谱峰的对称性都随着温度的升高而升高。随着柱温的升高,极性和非极性溶质的容量因子都减小,高效液相色谱系统压力和茶多酚的保留时间都呈减小趋势。同时,温度对分离度和有效塔板数都有影响,当温度和流速都增加时,柱效也得以增加,在30 ℃,流速为0.2 mL/min时,分析时间比25 ℃减少,分离效果比35 ℃好,峰形更窄且对称,充分说明了温度对柱效调节的有效性。当流速为0.2 mL/min时,色谱峰形好,且色谱柱能在15 min内分离所有的化合物。
2.3.4 检测波长的确定 试验采用200~400 nm全波长扫描,用3D色谱图分析发现,10种组分在210 nm处和278 nm处均有最大吸收,与目前已有的文献结果一致。对照目标物相应的2D谱图,10种标准物质在这个波长范围内,分离度好,峰高、峰型都能达到检测需要,但在梯度洗脱时,由于流动相的影响,造成实际样品在210 nm检测时基线漂移较明显,而在278 nm处,10种分析物灵敏度、分离度以及基线情况都很好,并且避免了溶剂的影响。因此,在试验过程中,采用278 nm作为检测波长。
2.4 标准曲线及线性范围
GA、CAF、GC、EGC、C、EGCG、EC、GCG、ECG、CG标准溶液浓度均在0.05~10.00 mg/L具有良好的线性关系,具体的线性方程及R2见表4。
2.5 检出限与定量限的确定
以3倍信噪比计算检出限(LOD),10倍信噪比测量定量限(LOQ),得到结果见表5。
2.6 方法的精确度和可选择性
2.6.1 方法的精确度 用外标法计算可以得出绿茶样品中的茶多酚浓度及其含量(表6)。利用本试验中建立的超高效液相色谱法检测绿茶样品中儿茶素、没食子酸和咖啡碱含量,进行精密度试验,结果表明,绿茶样品中各种特征物质含量的相对偏差RSD在1.77%~5.89%,说明该方法精密度良好。
2.6.2 方法的可选择性 进一步测定红茶和白茶样品中的儿茶素、没食子酸和咖啡碱含量,红茶中目标物含量在0~3.3%,相对偏差为0.28%~14.90%,白茶中目标物含量在0.05%~11.00%,相对偏差为0.51%~6.42%,该方法适用于红茶和白茶。儿茶素在每类茶中的含量差异较大,绿茶中的儿茶素含量最高,红茶中无GC和EC,白茶中EGCG含量最高,经过比较可以看出,绿茶是不发酵茶,儿茶素基本全部保留,白茶部分发酵,儿茶素有略微的分解降低,红茶的儿茶素在发酵过程中被大量的分解。
3 小结与讨论
本试验建立了超高效液相色谱法快速测定儿茶素、没食子酸和咖啡堿含量的方法,选用ACQUITY UPLC BEH C18(1.7 μm,2.1 mm×50 mm)色谱柱,柱温为30 ℃,流速为200 μL/min,流动相为0.1%甲酸水-0.1%甲酸甲醇溶液,检测波长为278 nm,该方法适用于检测绿茶样品中的特征物质。绿茶中多酚类占干物质总量的20%~30%,咖啡碱占2%~3%,其中儿茶素占多酚类总量的70%~80%。白茶所含有的功能性成分主要是茶多酚及其氧化产物、咖啡碱和氨基酸等,成品白茶的多酚类物质含量为18.3%~33.0%,儿茶素含量为7.84%~12.86%。红茶是发酵茶,茶多酚减少90%以上,生成茶黄素(TF)、茶红素(TR)和茶褐素(TB)等新成分。在制作时,绿茶不经过任何发酵过程,白茶经10.00%~30.00%程度的发酵,红茶的发酵度高达80%~90%[8]。红茶和白茶中存在其他目标物质,在后期试验中,可将试验方案进一步优化改进,以测定茶叶中更多的特征物质,使得该方法的选择性更大。
参考文献:
[1] 张静杰.基于代谢谱分析的工夫红茶加工工艺优化及其品质形成研究[D].合肥:安徽农业大学,2013.
[2] 谭和平,邹 燕,叶善蓉,等.茶叶中的多酚类物质及其分析方法综述[J].中国测试技术,2008(4):4-11.
[3] 吴 迪,杨丽珠,仇佩虹,等.茶中儿茶素的分离分析方法研究进展[J].分析科学学报,2008,24(4):468-472.
[4] 谢 果,何蓉蓉,栗原博.茶叶生物碱的生物合成与代谢的研究进展[J].中国天然药物,2010,8(2):153-160.
[5] 蔡明瑛,倪 辉,李利君,等.液相色谱检测乌龙茶水浸提物及其单宁酶水解产物中的儿茶素[J].中国食品学报,2013,13(5):211-219.
[6] 吕远平,何 强,姚 开,等.儿茶素提取的优化条件研究[J].四川大学学报(工程科学版),2004,36(2):65-68.
[7] 吴 娆.食用植物油多元掺伪鉴别技术研究[D].北京:中国农业科学院,2016.
[8] 刘 念.电化学技术在茶叶分类及重金属含量检测中的探索[D].长沙:中南大学,2014.
