系统药理学:中医药学研究新理论和新技术*
2019-01-29陈学通王永华
陈学通,粟 星,黄 超,李 燕,王永华,2**
(1.石河子大学新疆植物药资源利用教育部重点实验室 石河子 832000;2.西北大学生命科学与医学部 西安 710000)
1 引言
1个世纪以来,药物研发的主导范式是针对单一机制的单个靶标设计高选择性、高亲和力的药物。以此为目的,以数量巨大的化合物库为来源,针对明确的靶标进行体外高通量筛选,最终帮助科学家在较短的时间内筛选出针对特定药靶的高亲和力候选药物[1,2]。这些单靶标、高选择性药物在临床实践中成功得到了应用,例如磷酸二酯酶-5抑制剂(例如西地那非),α 1a肾上腺素受体拮抗剂(例如坦索罗辛),选择性环氧合酶-2抑制剂(例如塞来昔布)和激酶特异性抗肿瘤药(例如伊马替尼)等药物。证明了单靶标的高选择性药物确实能对疾病治疗起到效果。长期以来,受到西方药学的影响,中药学的大量研究也主要致力于从中药中寻找高活性、高亲和力小分子,期望借此阐明中药的药效学基础。虽然中药在临床上取得了显著的药效,却难以从中药中发现高活性、高亲和力的小分子。这一矛盾一直困扰着研究者。迫使我们对传统的药物研发模式进行反思,“一个疾病,一个靶标,一个药物”的哲学是否适用于所有疾病的治疗,高选择性、高亲和力为导向的药物研发策略是否具有普适性。
近年来,越来越多的研究表明药物的疗效与其靶点的亲和力高低并无必然的联系,某些低亲和力分子也同样具有良好的临床效果。例如阿司匹林。此外,近期研究发现,一个药物往往作用于多个靶标,并展示出较低的亲和力(对靶标的抑制常数大于10-6M)[3]。这表明一个药物的疗效通常与复杂的药理学相关联,并通过作用于多个靶标发挥药效[4,5]。而针对当前公布的药理学数据库的信息的统计分析发现,中药中存在大约有11万个具有多靶点、弱结合特性的小分子[6]。如何理解这些分子的治疗机制,并开发合适的药物筛选技术是研究者面对的主要难题。鉴于目前的认识水平和发展成果,为了重新认识弱结合分子的重要性以及作用机制,我们需要重新定义弱结合药物,并提出弱结合药物研究的新方法和新技术。
2 中药“弱结合-显效”理论
一直以来,中药弱结合分子作为药物的发现来源被忽视,其原因主要有三点。第一,目前许多科研人员的惯性思维仍然是药物与靶标需要紧密结合,他们认为弱结合药物对其靶标不具有特异性,从而会与多个非目标靶标发生相互作用[7]。第二,认为弱亲和力的分子对目标的结合量可能太低而无法推动反应。但是,如果局部浓度的弱结合剂足够高,它可以推动平衡,导致相当大的配体结合。第三,目前的药物发现方法无法筛选或者分析弱结合药物或者弱的生物相互作用,比如药物筛选时往往只保留分子库中抑制常数为1 μM或者更低的分子,弱结合分子在多种筛选过程中被轻易的剔除了[7]。这些因素导致研究者无法有效的在中药中发现弱结合药物。
幸运的是,我国药学家屠呦呦教授对青蒿素的研究促进了对弱结合药物的认识。2015年屠呦呦教授因为青蒿素获得了诺贝尔奖,随后引发了青蒿素的研究热潮。同年,新加坡国立大学和南京大学的研究人员用青蒿素类似物鉴定了124个与青蒿素共价结合的蛋白,指出青蒿素作用于多个靶标从而发挥疗效[8]。2016年,上海生命科学院的研究人员指出青蒿素可能通过结合多个靶标,调节多条细胞通路发挥协同抗癌疗效[9]。这些案例启发我们,在中药中存在着一类分子:在弱结合的情况下,可以通过作用于多个靶标达到治疗效果。
研究者在中药的研究中也发现了类似的现象。针对细胞炎症模型MAPK通路的4个关键靶点ERK、JNK、p38和MEK1/2,研究者系统地开展了药物抗炎和靶点结合力的关联研究(天然产物400个,西药分子200个)[10]。结合力评价发现:所有天然产物和上述4个靶点的结合自由能均大于-25 kcal·mol-1(弱结合力),而西药分子则普遍低于-40 kcal·mol-1(强结合力)。例如,木犀草素和上述靶标的IC50在29.6-300 μM之间,没有表现出明显的靶点选择性。而西药PD0325901和MEK1能够特异性结合,具有很强的亲和力(IC50=0.33 nM)。然而,令人惊讶的是,所有分子中木犀草素的抗炎效果最佳。进一步研究发现,木犀草素的3个靶点(JNK+p38和MEK1/2+p38)呈现“并联协同”作用,当其协同增效倍数超过一定阈值(CI<0.9),即可激发显著的生物学表型效应(图1)[10]。此外,研究者在迷迭香酸研究中拓展了该发现,尽管该分子和 3个作用靶点 ACE,PTGS2,REN(IC50=30-500 μM)呈现弱结合效应,但是由于靶点之间的“串联协同”,使其达到较好的血管舒张作用[11]。更加重要地,在复方热毒宁注射液研究中,发现了其3个主要活性成分京尼平、东莨菪亭和绿原酸在采用和注射液同等含量联用时,具有与注射液相当的抗炎效果,而3个成分单独使用(与注射液同等含量)则几乎没有抗炎效果[12]。靶点研究表明:这3个成分分别靶向MAPK通路上下游的紧密联系的3个靶点,即JNK,ERK,c-Jun(IC50均 >300 μM),在这条通路上激发了很强的“串联和并联协同”作用,形成了“多分子接力”较强抗炎效果。
以上这些发现给予研究者巨大启发。由于人体内复杂的生网络,当单靶点、强结合药物在面对复杂疾病时(例如肿瘤、心血管病、阿尔兹海默症等),无法取得预期的疗效,而靶向多个靶点的弱结合药物通过“串联和并联协同”等作用站展现出更优秀的治疗效果。由此我们提出了中药“弱结合-显效”理论:如果一个中药单体结合于多个靶点且亲和力均较低,当其作用的靶点之间满足特定的拓扑结构和网络动力学条件时,可产生串联或并联协同作用,从而激发显著的表型效应。这一理论也可以拓展到复方中多分子互作研究。
3 中药“弱结合-显效”研究评价新技术和新方法
为了重新认识中药“弱结合-显效”的重要性并理解其作用机制,我们需要在蛋白组尺度上定量和系统分析药物-靶标相互作用,并预测药物的表型反应,从而在浩瀚的中药成分库中筛选出符合“弱结合-显效”理论的潜在药物。2007年,英国Dundee大学药理学家Hopkins首次提出了网络药理学概念(Hopkins 2007),对药物研发的理念、策略和方法产生了深刻的影响。科学家逐渐开始从生物网络平衡的角度阐释疾病发生过程、认识药物与机体的相互作用并指导新药发现。促进了网络药理学、多向药理学、系统药理学等新兴学科蓬勃发展,为多靶标弱结合药物的开发奠定了基础。
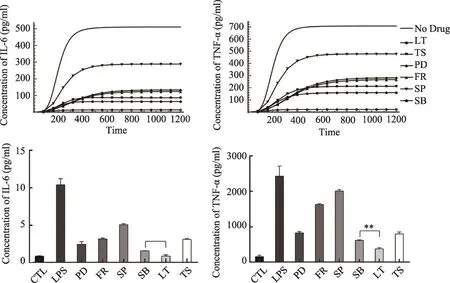
图1 (A)模拟两个天然产物木犀草素(LT)和丹参酮IIA(TS)以及选择性抑制剂PD(MEK抑制剂),FR(ERK抑制剂),SP(JNK抑制剂),SB(p38抑制剂)在10 μM浓度时对IL-6和TNF-α生成的抑制效果。这些化合物在10 μM时对靶标的抑制率由他们的IC50曲线推测得到。(B)细胞实验测定的两个天然产物和四个选择性抑制剂在10 μM时对THP-1细胞产生IL-6和TNF-α的抑制效果
3.1 实验测定弱结合药物
考虑目前的实验筛选方法,筛选弱结合药物仍然是一项挑战。一般情况,弱结合药物或弱的生物相互作用由于筛选上和分析上的困难很少被研究。许多高通量筛选(HTS)试验依赖于直接检测方法,例如荧光反应,吸光度或活性率,这可能成为评价弱结合的障碍。由于实验设计的局限性,高通量筛选过程会产生假阳性和阴性结果,尤其是评价存在弱结合化合物时。尽管如此,基于酶活抑制试验的高通量筛选如果设计合理,仍可以检测到半数抑制浓度(IC50)小于10-4M的弱结合化合物[13]。此外,还有一些可以筛选弱结合化合物的潜在方法,例如核磁共振(NMR)[14],质谱(MS)[15],X-射线晶体照相术[16],亲和色谱[17],毛细管电泳[18]和表面等离子体共振[19]。这些方法在筛选弱结合药物方面具有一定的实用性。但是需要消耗巨大的资源,无法高效的从中药巨大的成分库中筛选出合适的药物。我们需要推出新方法和新技术以解决当前弱结合药物筛选中遇到的困难。
3.2 计算方法筛选弱结合药物
界定和预测弱结合药物的多向药理学效果需要定量的理解其靶标的结构和功能,还需要了解靶标和小分子在生物网络环境下的相互作用[20]。此外细胞成分之间也通过类似网络的方式发生相互作用进而行使功能[21]。网络结构本身的动力学属性很大程度上决定了互作分子的生物功能,因此与药靶有关的生物网络的结构可能帮助揭示弱结合药物的作用模式[22,23]。许多疾病相关通路的数学模型已经被开发出来,具有阐明疾病机制和识别有效治疗策略的潜力[24,25]。分析这些疾病相关分子网络的性质可以发现潜在药靶并理解他们之间的互作模式[26,27]。例如,一些特殊的信号元素,如PI3K,Akt和胰岛素受体底物家族[28-30],是多条信号通路的重要连接点,已被作为药物开发中的重要靶标。模拟网络行为表明部分抑制多个靶标比完全抑制单个靶标更为有效[5]。此外,为了定量分析药-靶互作,可以从刻画药-靶互作的热力学和动力学性质开始[31],然后鉴定靶标被药物结合后的构象和化学状态[32]。这个过程主要依靠配体-蛋白的分子对接(docking)[33],配体-蛋白复合体的自由能计算和分子动力学模拟[34,35]。这些计算的方法为我们筛选弱结合药物提供了一定的思路,但是当前并没有一个综合的方法来确定中药中的弱结合药物,我们需要提出一个综合性的方法和框架来解决弱结合药物的筛选问题。
4 基于系统药理学的弱结合分子筛选新方法和新技术

图2 网络基元动力学分析技术流程图
为了探索药理学新领域并合理设计弱结合药物,有必要将那些相对独立的计算和实验技术整合到一个框架下面。针对目前筛选有效的多靶标弱结合药物过程中存在的问题,研究者有机的整合通路及网络分析,蛋白组范围预测药物-靶标相互作用以及药代动力学模型,开发出一种用于寻找多靶标弱结合药物的系统方法:基元模块动力学分析(ESDA:Elementary subgraphs dynamics analysis)方法[10]。该方法,可以有效的预测细胞信号网络对多点弱扰动的响应,随后根据网络的拓扑性质选择合适的靶标组合,利用系统药理学方法预测与靶标组合发生互作的化合物并实验测定其结合力以及药效(图2)。首先,对一系列基元模块进行动力学模拟来研究在多靶药物作用下的网络结构和动力学参数。其次,将基元模块应用到经典的MAPK通路寻找最优靶标组合。再次基于这些靶标组合,利用系统药理学方法,从巨大中药分子库中筛选候选的弱结合药物。然后,采用分子动力学模拟和结合自由能计算来评价化合物与靶标的亲和力,用激酶抑制试验验证预测结果。最后,在体外实验验证了这些多靶标弱结合药物候选物的潜在疗效。
4.1 弱结合成分药物靶标预测
药物在经过有机体的吸收、分布、代谢和排泄(ADME)过程后,保留在体内的活性成分需要靶向生物大分子(DNA、RNA和蛋白质)来发挥药效,因此药物分子的靶标识别是药物开发的关键。然而目前,只有化学药物和少部分的天然产物具有靶标信息,大部分天然产物尤其是具有多靶标属性的天然产物其靶标仍然不清楚,这显然阻碍了我们开发多靶标弱结合药物的进程。针对此问题,我们创新性的开发了靶点预测技术(SysDT)直接根据小分子的结构信息和靶点的一级序列信息来获得小分子与靶标的互作关系[36]。同时鉴于蛋白的一级序列无法全面反映其与分子的互作信息,进一步利用开发的加权系综相似度算法(WES)预测天然产物的靶标[37]。最后,利用药-靶激活/抑制模式分析模型(PreAM)预测化合物-靶标的互作关系[38]。
靶点预测技术(SysDT)是一个大规模整合药物、基因组和药理学数据信息,借助人工智能技术实现了准确关联药物结构特征和药理学表型的多重药-靶互作分析技术:1)以DrugBank数据库(http://drugbank.ca/)构建了一个包含6511药物和3987个靶点的原始药物-靶点关系数据;2)分别用Dragon程序(http://www.talete.mi.it/index.htm)和 ProFeat web sever(http://jing.cz3.nus.edu.sg/cgi-bin/prof/prof.cgi)计算分子述符和蛋白描述符;3)随后综合利用随机森林(Random Forest,RF)和支持向量机方法(Support Vector Ma-chine,SVM)建立了一个准确关联药物结构特征和靶点药理学表型的多重药-靶互作分析技术。该方法可以直接根据分子的结构特征预测出其潜在的靶标。
加权系综相似度算法(WES),通过挖掘特定配体群的系综特征,结合生理参数,依靠贝叶斯网络整合技术,实现了蛋白分子互作的准确预测(>79%):从BindingDB数据库中得到包含了inhibitory(Ki)和IC50信息的98327个蛋白-配体关系,其中包括1788个蛋白和68777个分子;利用CDK程序和Dragon程序计算分子描述符;识别出配体中与药理学信息相关的关键结构特征;通过评估总体相似度(整体)而不是单一的配体判断来确定分子和靶标的亲和性;通过贝叶斯网络和多变量核心方法整合标准化综合相似度(Z score)进行分子靶标预测。
药-靶激活/抑制模式分析模型(PreAM)是运用超平面分割技术来提取靶点蛋白中参与决定互作模式的结构因子,并整合化合物药效团特征的新策略构建的一个高准确率的药物激活/抑制算法:来自DrugBank数据库的获得6006个已知激活/抑制信息的药物-靶标关系,其中包括1251个激活关系和4755个抑制关系;分别用Dragon程序(http://www.talete.mi.it/index.htm)和 ProFeat web sever(http://jing.cz3.nus.edu.sg/cgi-bin/prof/prof.cgi)计算分子述符和蛋白描述符;利用随机森林(Random Forest,RF)的方法训练出一个药物-靶标的激活/抑制信息分类器并用于新化合物-靶标互作关系的预测。
综合上述3个模型预测出化合物的潜在的靶标,并为后续的多靶标弱结合药物筛选奠定基础。
4.2 弱结合药物多靶点扰动模拟
针对特定的疾病机制开发多靶标弱结合药物,首先需要了解该疾病机制所涉及的功能蛋白或信号分子等要素,同时需要了解这些要素所处的复杂网络环境。细胞网络的平衡原则使得细胞对分子扰动具有弹性的同时又具有对微妙的输入信号的敏感性。洞悉这种机制或许能够利用细胞的控制回路加快多靶标弱结合药物的开发进程,获得药效更好、剂量更低和耐药机率更少的药物[39]。在生物网络中,绝大部分成分都不是孤立的,或多或少的与其他成分具有关联,相互促进或者制约,使得系统处于动态的平衡。但不论网络多么复杂,可能只存在为数不多网络拓扑结构能够有力的执行任何特定的生物学功能,那些存在较少参数约束的拓扑结构可能更为有利。根据生化细节和进化历史,相同的网络拓扑结构核心组可能是细胞行为的基础[40,41]。因此,在面对复杂的、动态的生物网络,有必要对其进行简化,抽提出核心组件方便我们进行分析和运用。当前,一些科学家利用少数节点来简化复杂的网络结构,并模拟外界因素对这种简化网络的干扰,归纳总结了一些基本的网络结构单元并研究他们的拓扑学和动力学特性[42-45]。
由此,我们在前人研究工作的基础上进行总结和扩展,构建了一系列三成份的基元模块(elementary subgraphs),基本包含了3个成份之间所有可能的互作方式。随后用数学模型来模拟这些基元模块动态行为,帮助我们更清晰的理解他们的结构特征。最终,我们提取出了33种复杂网络中的核心组件(基元模块):由两个靶标和一个下游效应器构成的三成分拓扑结构。根据这些拓扑结构的特点进行了分类,按照结构分成了串联模块和并联模块两大类,包括11个串联模块和22个并联模块。随后运用常微分方程对这些基元模块进行了模型构建,并假定存在两种靶标抑制剂对基元模块进行扰动。模拟这些基元模块在多点扰动下的响应情况来探索哪些基元模块存在协同效应,而哪些基元模块是拮抗效应,并且探索动力学参数对这些协同/拮抗模块的影响。最终,我们根据模拟结果将基元模块分为18个非参数依赖模块和15个参数依赖模块(图3)。这些模块为理解复杂网络的基本特征提供了一种方法,并为筛选弱结合药物的网络拓扑特征奠定了基础。
4.3 弱结合药物最优靶标组合
针对一个特定的分子网络开发多靶标药物,首先要面对拓扑结构复杂,动力学参数多变,多途径之间反馈、串扰等困难。在没有弄清楚分子网络的特征之前,针对网络中的若干节点开发多靶标药物是盲目的。原因在于分子网络的鲁棒性和冗余性,不恰当的扰动靶标组合往往得不到有效的系统响应,甚至产生不利影响。开发多靶标弱结合药物首先要解决的就是靶标组合的选择问题,基于优化的靶标进行药物筛选不仅能够获得具有良好药效的候选化合物,还能够避免产生耐药性等问题。
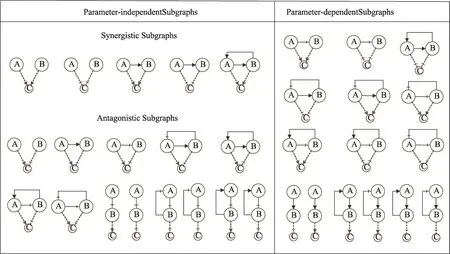
图3 对33个基元模块的进行动力学模拟分析协同效果
信号转导通路是众多生物反应通路中的一种,是生物体最基本也最重要的组成要素,几乎所有的生命现象都与细胞内信号转导有关。而信号转导的失调与多种疾病的发生发展具有直接或间接的关系[46]。了解信号通路的特征有利于我们有目的性的开发多靶标弱结合药物。在众多的细胞内信号中,MAPK信号通路是一个在进化上相当保守并且被广泛研究的信号通路,它在调节机体对应激和炎症的响应方面扮演着重要角色[47,48]。因此,我们利用前人实验获得的和自己估计的动力学参数,通过常微分方程构建了浓缩的MAPK信号通路模型(图4)。并基于网络基元模块理论,在这个真实的生物网络中探寻符合非参数依赖或参数依赖的基元模块以获得最优靶标组合。简单来说,我们首选选取了该信号通路中的经典药靶进行组合扰动,以两种炎性因子的生成量来评估系统响应情况。扰动模拟包括了对单靶标的强扰动,对串联模块靶标的弱扰动以及对并联模块的弱扰动。最后根据下游炎性因子生成受到抑制的程度选择合理的靶标组合,发现对3个串联模块多点弱扰动后,只有组合抑制MKK3/6和p38表现出对IL-6和TNF-α理想的抑制效果,而对于12个并联模块,同时抑制ERK和p38表现出对IL-6和TNF-α最好的抑制效果。此外,多点弱扰动的5个最优靶标组合中都含有p38,表明针对MAPK信号通路开发多靶标弱结合抗炎药时,优先考虑含有p38的靶标组合。
4.4 弱结合药物筛选与验证
当前的实验手段筛选多靶标弱结合化合物存在很大挑战,用计算模拟的方法作为辅助手段可以降低多靶标弱结合药物筛选的难度,节约时间和提高成功率。为此。当前,我们利用系统药理学方法中的靶标预测模型和互作类型预测模型对中药中全部小分子进行了靶标和互作方式预测,根据MAPK信号通路靶标组合反向筛选获得了32个多靶标化合物,这些化合物大部分被报道具有抗炎活性,一定程度上说明我们的前期的工作的可靠性。其次我们采用分子动力学模拟以及结合自由能计算的方法初步评价候选化合物与靶标的亲和力。随后选取木犀草素与丹参酮IIA作为多靶标弱结合化合物,以几种激酶抑制剂作为单靶标强结合化合物对照,进行了体外实验验证。结果表明,在酶学水平上木犀草素和丹参酮IIA对其靶标的IC50在微摩尔浓度范围,远大于现有的激酶抑制剂,但是在细胞抗炎实验中显示出与激酶抑制剂相当甚至更好的药效。
综上所述,我们系统地分析了网络基元模块的性质,并利用基元模块选取信号通路中的最优靶标组合,随后基于系统药理学方法筛选出多靶标弱结合药物。这些结果,表明基于系统药理学筛选多靶标弱结合药物是一种切实可行的方法,可以为新药开发、老药新用,天然产物的开发利用提供新思路。

图4 数学模拟MAPK信号通路
5 中药“弱结合-显效”理论和方法在活血化瘀机制的验证和应用
活血化瘀是中医的一个重要理论和治疗原则。研究者以典型血瘀型冠心病为切入点,着眼于“弱结合-显效”这一新视角,从中药作用靶点,起效模式等方面出发,以川芎、丹参和三七典型活血化瘀中药为例开展研究。发现了这些草药共同作用于冠心病相关靶点13个且结合力均较弱(IC50>300 μM)。例如川芎内酯(ligustilide)与PTGS2,GABRA1串联协同抗血栓形成;丹参素A(salvianic acid A)与蛋白凝血因子Xa(F10),ADRB1,PPARγ和NOSs结合,并联协同促进血管舒张[49]。这种“弱结合-显效”模式在银杏叶治疗中风的活血化瘀机制中得到进一步证明[50]。此外,基于“弱结合-显效”理论和方法,研究者阐明了红花和山楂等单味药治疗CVD效果不显著的原因:其主要活性成分黄杉素、丁香脂素、原紫草酸等与CVD的主要靶点ERB,PPAR为弱结合(>500 μM)且不符合显效协同模式。结合前期构建的CVD靶点数据库(CVDSP,http://lsp.nwu.edu.cn/cvdsp.php),发现了丹参酮VI,黄杉素和丁香脂素3个分子的靶点满足协同增效的拓扑结构和动力学条件,由此确定了一个治疗缺血型冠心病最优组合:“红山丹”(红花+山楂+丹参,最佳质量比为1∶1∶2),在动物实验上达到和同类西药相当的疗效[51]。
6 结论与展望
开发高亲和力的选择性配体是过去几十年药物开发的主导方式。然而由强效的先导化合物获得的药物候选物往往在药物开发过程中具有较高的失败风险率。同时,分析市售的高亲和力药物和天然产物表明疗效与高亲和力并无必然联系。从经典的蛋白折叠到近来代谢、基因调控和信号转导的最新发现,都表明在分子水平上,弱的相互作用在生物系统的分子识别中扮演着重要角色。以网络生物学的观点来看,弱相互作用有利于生物网络的鲁棒性和多样性,因此集体的弱相互作用可能比单一的强相互作用对生物系统更具深刻的影响。
设计有效多靶标弱结合药物的另一个关键问题或许是在复杂细胞网络中寻找合适的靶标组合。尽管实验工具例如高通量筛选有希望发现弱结合药物,但通常是耗费时间且效率低下,而且呈指数增长的潜在药物靶标组合也使实验工具无法满足需要。合适的计算模型和算法以及丰富的数据库资源,能有效的减少寻找潜在靶标组合的搜寻空间。因此,系统的整合计算工具与实验策略可以有效识别弱结合化合物,实现弱结合药物在治疗不同疾病表型上的全部潜力。
天然产物和他们的组合通常作用于多种药物靶标,这些靶标包括并超出了目前FDA批准药物的有限靶标空间,因此具有相当大的治疗潜力。从这些天然材料里获取的弱结合药物真正有机会在发挥最大疗效的同时最大限度的降低副作用。我们提出了一种可靠的方法来识别多靶标弱结合化合物。由于这种方法可以作为高亲和力药物研发的补充,我们可以想象更多的疗法将会被重新发现。因此,如果更多的科学家开发药物时不单是考虑紧密结合到蛋白上的化合物,同时也关注弱结合或者瞬时结合到多个靶标的小分子,新的药物开发将具有充满希望的美好未来。
