14个耳聋家庭的临床特征及遗传学病因研究△
2019-01-24赵军孙菲菲施健唐艳吴笛邱金红朱爱华张鲁平
赵军 孙菲菲 施健 唐艳 吴笛 邱金红 朱爱华 张鲁平
耳聋是临床上常见的遗传病之一,其中50%以上由遗传因素所导致,即遗传性聋[1]。遗传性聋具有高度异质性,至今已有130余个耳聋基因被克隆或鉴定(http://hereditaryhearingloss.org)。如何快速精准完成耳聋患者的基因诊断,特别是为婴幼儿耳聋患者明确病因,为耳聋家庭提供行之有效的精准化预防和诊治方案,是临床工作中的一大挑战和难点。本研究以耳聋家庭为单位,使用两步法模式(微阵列芯片法+高通量基因捕获技术方法),明确了14个耳聋家庭的遗传病因,结果报道如下。
1 资料与方法
1.1研究对象 本研究14个耳聋家庭的14例先证者皆因双侧不同程度的感音神经性聋就诊于南通大学附属医院耳科门诊(2017年3月至2018年4月),其中12个家庭的父母听力正常,2个家庭的父母为聋哑人,均为非近亲结婚。对14个家庭进行了详细的病史采集包括:听力障碍的起始年龄及诱因、进展情况、有无耳鸣、眩晕等伴随症状、既往有无系统性疾病、耳毒性药物应用史及噪声接触史等,进行全身体检排除综合征型遗传性聋,并进行听力学检查及颞骨高分辨率CT扫描。本研究得到受试者的知情同意及南通大学附属医院伦理委员会伦理论证认可。
1.2听力学检查及听力学表型的分析 对纳入研究的耳聋患者进行如下检查:①纯音听阈检测(适用于配合良好、大于4岁的患者);②声导抗检查;③畸变产物耳声发射 (DPOAE)检测;④听性脑干反应(ABR)及听性稳态反应(auditory steady-state responses, ASSR)测试, 用于年龄≤4岁患者;综合上述检查确定听力损失程度。以较好耳0.5、1.0、2.0及4.0 kHz四个频率的平均听阈判断听力损失程度: 20~40 dB HL为轻度听力损失;41~70 dB HL为中度,71~95 dB HL为重度;>95 dB HL为极重度。根据家系调查和听力结果绘制系谱图。
1.3候选致病基因突变筛查 家系成员均抽取外周静脉血2 ml (采血管含EDTAK2抗凝剂),提取基因组DNA备用。实验方法同参考文献[2,3]:①采用晶芯15项遗传性聋基因检测试剂盒(微阵列芯片法),检测样本4个基因的15个常见突变位点:GJB2(35delG、235delC、176del16、299delAT)、 SLC26A4(2168A>G、919-2A>G、1174A>T、1226G>A、1229C>T、1975G>C、2027T>A、IVS15+5 G>A)、线粒体12S rRNA(1555A>G、1494C>T)和GJB3(538C>T)。将基因芯片未能确诊的阴性突变患者,通过北京迈基诺基因科技有限责任公司开发的耳聋芯片,定向捕获所有已知的及相关的耳聋基因的外显子及其侧翼内含子序列,经二代测序数据分析后得到疑似致病基因突变位点;②常规Sanger测序方法验证;③验证突变位点在家庭成员中与耳聋表型的共分离情况。
2 结果
2.114个家庭耳聋患者的诊断及干预 本研究14个家庭共51人,20人耳聋,系谱分析均符合常染色体隐性遗传特征。经详细的病史询问及体格检查,确诊的20例感音神经性聋患者,年龄6个月~72岁,听力损失从中度、重度到极重度不等(表1),双侧基本对称; 5例自诉有耳鸣病史。20例耳聋患者其他系统均未见异常,无耳毒性药物接触史和噪声接触史,无新生儿期听力损失高危因素及智力障碍。14例先证者(HM1~14)均行颞骨高分辨率CT扫描,10个家庭的先证者(HM1~9、14)未发现中耳、内耳结构异常及内听道的占位病变,4个家庭的先证者(HM10~13)证实有双侧前庭水管扩大。其中6例先证者已行单侧人工耳蜗植入术,HM1、7、10~12家庭的5个患儿开机后对声音反应好,术后都不到一年,目前在进行听觉言语康复;HM13家庭的患者自幼佩戴助听器,双侧听力损失渐进性加剧,14岁时发展至双侧极重度聋,行右侧人工耳蜗植入术,目前该患者能够进行正常的言语交流。HM5、8、9先证者佩戴助听器,其中HM8先证者自幼佩戴助听器,言语发育尚可,能进行简单的日常会话,自诉有耳鸣且双侧听力损失近年来有加重趋势。
2.2候选致病基因筛查结果 14例先证者采用十五项遗传性聋基因诊断芯片检测结果见表1,①明确了8例先证者的遗传学诊断,其中6例为GJB2双等位基因突变,2例为SLC264双等位基因突变;8例后续经常规Sanger测序方法验证,准确率达到100%;②检测到1例GJB2、2例SLC264单等位基因突变;③3例未能检测出突变位点。
对6例未能确诊的先证者,进一步使用定向捕获联合二代测序技术进行耳聋基因检测,结果如下:①GJB2双等位基因突变3例:HM5家庭的先证者为c.235delC/c.380G>T,HM6家庭为c.109G>A/c.571T>C(图1),HM9家庭双基因致病为GJB2 c.109G>A纯合+TBC1D24 c.1156T>C杂合(图2);②SLC26A4双等位基因突变2例:HM12家庭的先证者是 c.919-2A>G/c.2167C>G(图3); HM13家庭为c.626G>A/c.2168A>G;③HM14家庭的先证者为相对少见的耳聋基因MYO15A突变:c.6956+9C>G/c.10245_10247delCTC(图4)。除HM5、6两个家庭外,其余12个家庭先证者的双等位基因变异经常规Sanger测序方法验证,均分别来自于父母亲。
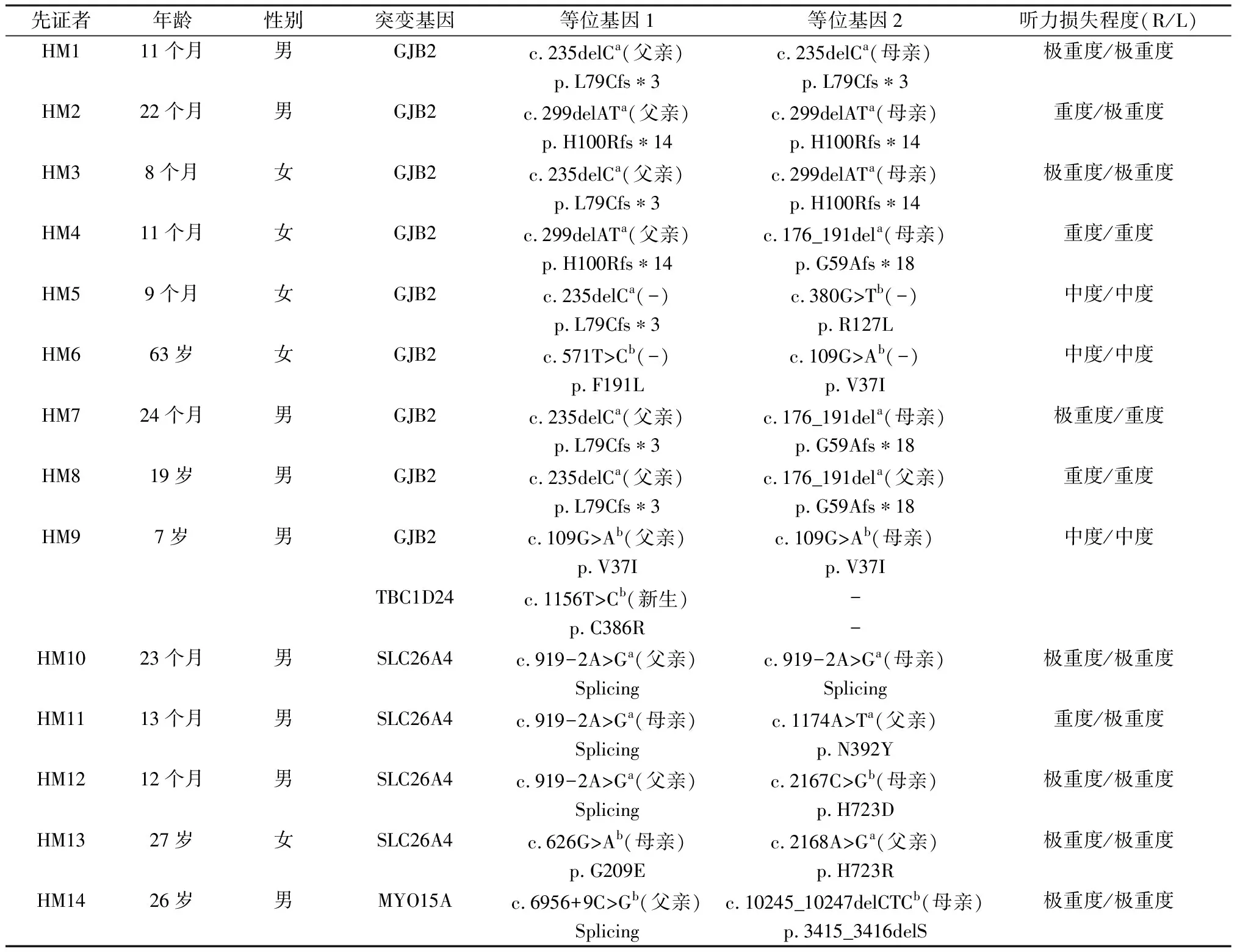
表1 14个耳聋家庭先证者年龄、性别、突变基因、突变位点及听力损失程度
注:a:芯片检出的突变;b:二代测序检出的突变;(-):未能采集到父母双亲的血样,不能明确突变的来源;Splicing:剪切突变; *:移码后遇到终止密码子时会用*号提示终止密码子的位置,如p.L79Cfs*3表示碱基缺失发生移码,后面的碱基补上来开始进行编码,编码到第3个氨基酸的时候是终止密码子。R/L:右耳/左耳
3 讨论
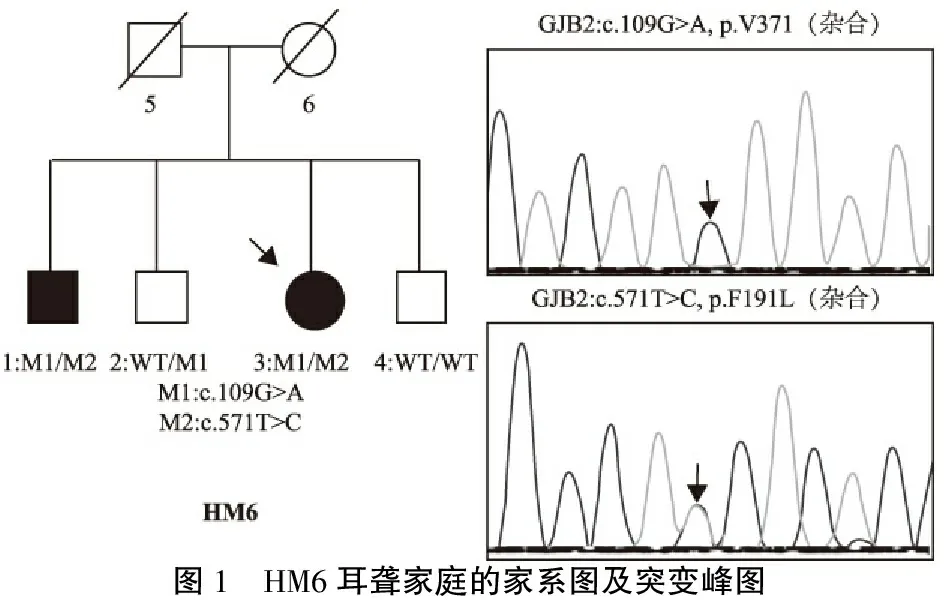
图1 HM6耳聋家庭的家系图及突变峰图
本研究14个耳聋家庭中,9个家庭明确为GJB2双等位基因突变致病,其中8个家庭先证者的听力学表型都为先天性聋或语前聋,只有一个家庭先证者表现为语后聋或渐进性听力下降;且3个家庭的基因型和听力学表型至今都无报道:①HM5家庭先证者的基因型为复合杂合变异c.235delC/c.380G>T,在国内,尤其是本研究所在的地区,c.235delC是GJB2最常见的致病变异位点[3,4],而国内外尚无c.380G>T(p.R127L)变异引起耳聋的病例报道。最近有研究证实p.R127L具有致病性[5],HM5家庭的先证者已有9月龄,为双侧中度感音神经性聋,进一步证实了p.R127L为致病性变异。HM5家庭的先证者为先天性聋哑,但其父母拒绝基因检测,故不能明确其父母是否同为GJB2基因突变的同证结婚。②HM6家庭先证者的突变为c.109G>A/c.571T>C,该家系有两人患病,起病年龄都在35岁以后,渐进性加剧,都伴有耳鸣。目前已明确c.109G>A(p.V37I)是致病变异[6];而对于c.571T>C(F191L),Ambros等[7]进行GJB2不同突变体的体外功能表达研究发现:F191L突变体可导致GJB2编码的缝隙连接蛋白Cx-26的运输功能异常。由于F191L为罕见变异,文献中未见F191L纯合和复合杂合突变致聋的病例报道,故对于F191L是否为致病性变异,目前尚无定论。本研究发现F191L高度保守、生物学分析提示该变异具有致病性、在家庭HM6共分离,同时结合Ambros的体外功能实验,高度提示F191L为致病性变异。③HM9家庭的先证者很可能为双基因致病:GJB2 (c.109G>A, p.V37I纯合) + TBC1D24(p.C386R,杂合)。p.V37I纯合变异大都引起迟发性轻度-中度的语后聋,随年龄增长渐进性加剧[3,8];但本研究中HM9家庭的先证者目前仅7岁,为先天性中度语前聋。Chai等[8]曾发现一例由GJB2 (p.V37I)纯合突变+CDH23 (p.Y1995X)纯合变异引起的极重度耳聋患儿;与Chai的报告类似,本研究中的该例先证者携带双基因变异;不同的是HM9的先证者携带的另外一个耳聋基因是TBC1D24,本研究团队先前已证实一常染色体显性遗传的非综合征型聋大家系的致病基因及其突变位点是TBC1D24杂合突变p.S178L[9]。HM9的先证者携带的TBC1D24杂合突变p.C386R是新生变异,该突变位点高度保守、生物学分析高度提示该变异具有致病性,结合先前的报告,推测HM9的先证者可能是双基因变异导致的中度聋(图2)。
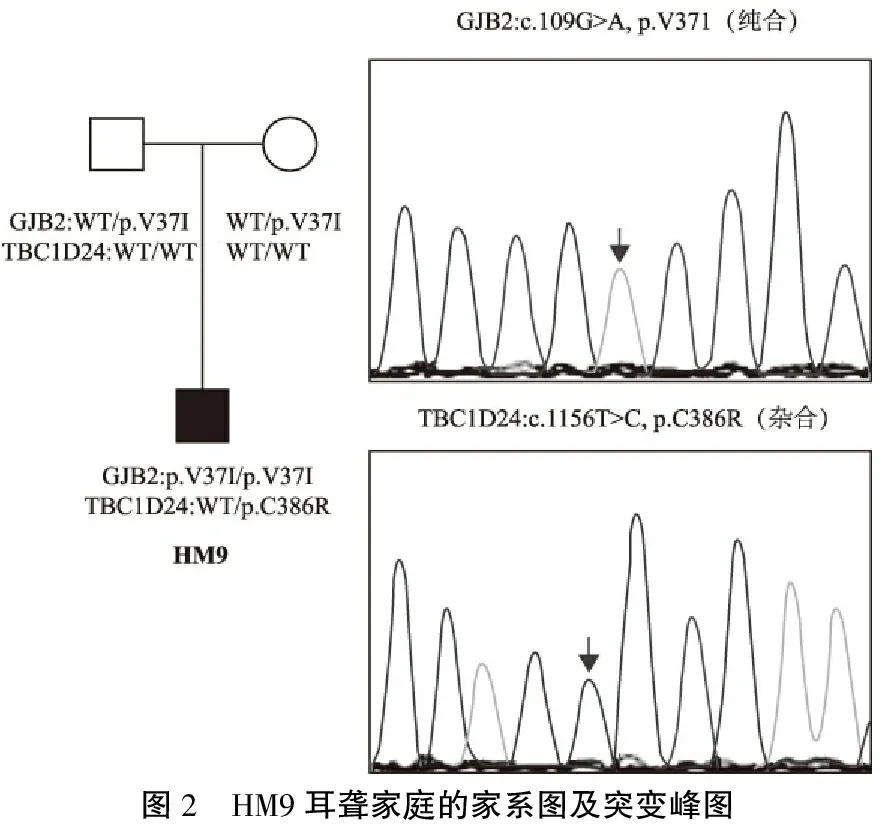
图2 HM9耳聋家庭的家系图及突变峰图
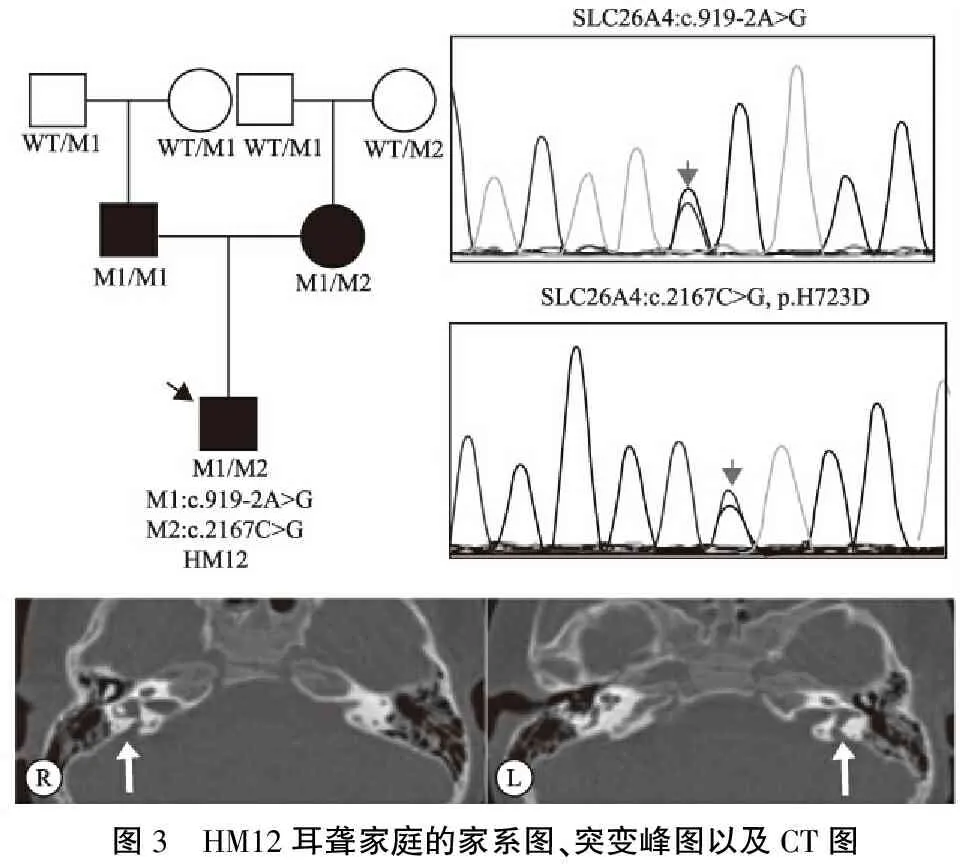
图3 HM12耳聋家庭的家系图、突变峰图以及CT图
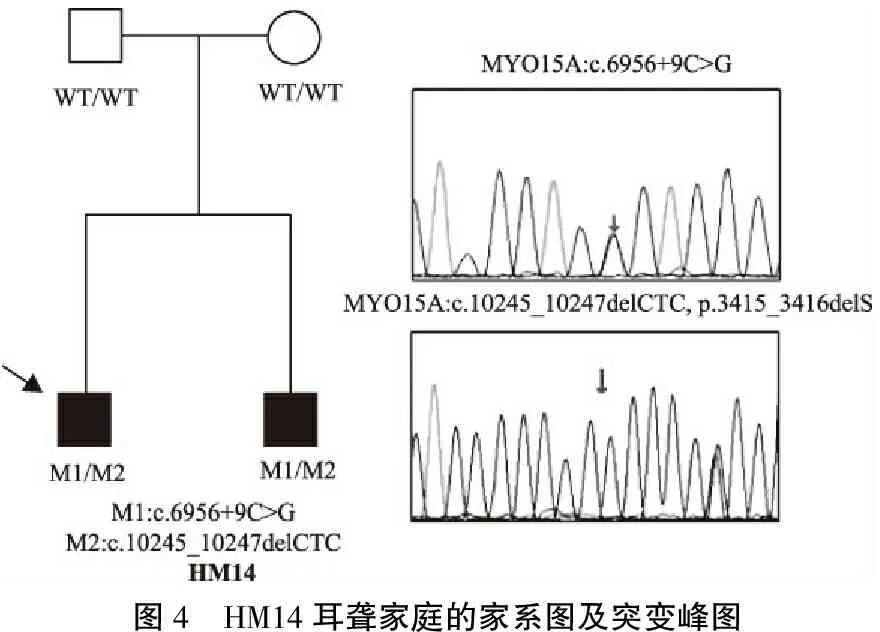
图4 HM14耳聋家庭的家系图及突变峰图
SLC26A4基因的突变范围几乎涉及该基因所有编码区域,除了少数的热点变异,尚有近200种相对少见的变异位点,包括大片段的缺失[3,10,11],本研究的两步法策略成功明确了4个耳聋家庭为SLC26A4基因变异致病。值得一提的是,HM12、13两个家庭从正反两方面阐明了先天性聋一级预防的重要性、迫切性及其现实意义[12]:①HM12家庭先证者的父母亲均为大前庭水管综合征导致的先天性聋哑,属于同一耳聋基因变异的聋人与聋人相结合的同证婚配(图3),所以理论上他们的后代百分之百是先天性耳聋患者;如果他们婚前能够明确基因诊断,进行遗传咨询,可以减少聋儿的出生;② HM13家庭的先证者携带复合杂合突变c.626G>A/c.2168A>G,其未婚夫听力正常,却携带杂合c.919-2A>G,对这个家庭进行精准的耳聋遗传咨询及产前指导,可以有效避免聋儿后代的出生。
本研究中,对于十五项遗传性聋基因芯片检测结果为单等位基因变异或者阴性的家庭,通过高通量基因捕获技术做了进一步的耳聋基因检测:一方面发现了上述常见耳聋基因罕见的突变位点;另一方面还发现了相对少见的耳聋基因及其变异:耳聋芯片检测阴性的家庭HM14明确为MYO15A基因变异(c.6956+9C>G/c.10245_10247delCTC);在韩国的常染色体隐性非综合征型聋患者中MYO15A基因是仅次于GJB2、SLC26A4及CDH23的第四大致聋基因[13];在我国也有报道,MYO15A基因突变引起的耳聋,均为双侧重度-极重度感音神经聋[14~17]。文中HM14家庭的两例患者都为聋哑人,为双侧先天性极重度感音神经性聋,首先,尽管该复合杂合突变至今无报道,但是上述两个变异在既往文献中均有报道, c.6956+9C>G属于移码突变[14],c.10245_10247delCTC属于小片段缺失,导致3416位少了一个丝氨酸(S)[13];其次该突变在HM14家庭共分离;且未发现其他可疑的致病变异,与MYO15A的隐性遗传方式一致。以上基本明确了HM14家庭的遗传病因,同时该家庭的两例耳聋患者已到婚育年龄,今后从重点怀疑和高危人群耳聋基因筛查的一级预防着手,可以从根本上阻断该家庭先天性耳聋的遗传。
目前,耳聋基因检测有多种方法[3,9,18,19],本研究改进了先前的检测策略[3],以每个耳聋家庭为单位,使用两步法成功鉴定出14个耳聋家庭的致聋基因及其突变位点,先使用十五项遗传性聋基因诊断芯片成功鉴定出8个耳聋家庭的遗传病因,(GJB2变异引6个,SLC26A4 突变2个,检出率为(57%,8/14),这8例后续经常规Sanger测序方法验证,准确率达到100%。耳聋基因芯片技术快速、操作简便、检测成本低、检出率高及准确率高,应结合本地区耳聋群体中基因突变谱的特点[3],首选基因芯片技术进行耳聋基因检测。对于上述检测不能明确基因诊断的,再以家庭为单位,应用高通量测序技术,对每个耳聋家庭的患者及正常对照者携带的变异信息进行甄别,从候选的变异信息中筛选出真正的致病变异,最终找出与耳聋表型共分离的基因变异。
综上所述,本研究以每个耳聋家庭为单位的二步法基因检测模式,成功鉴定出14个非综合征型聋家庭的致病基因及其突变位点,丰富了本地区的遗传性耳聋基因突变谱,获得了上述突变基因型相对应听力学表型的宝贵临床资料,同时为这些耳聋家庭提供了个体化精准服务,取得了良好的经济及社会效益,值得推广和应用。
