猪流行性腹泻病毒S蛋白诱导内质网应激的研究
2019-01-22马艳龙尹灵丹郭珊珊刘平黄
马艳龙,薛 美,符 芳,尹灵丹,郭珊珊,冯 力,刘平黄
(中国农业科学院 哈尔滨兽医研究所, 黑龙江 哈尔滨 150069)
猪流行性腹泻(Porcine epidemic diarrhea,PED)是由PED病毒(PEDV)引起的一种急性接触性肠道传染病。该病毒主要损伤猪的肠道黏膜,临床上表现为水样腹泻、脱水、呕吐为主要特征的急性肠炎。各个年龄段的猪均对PEDV易感,对一周龄以内的仔猪危害最大,感染率和死亡率均在80%以上,给我国乃至世界养猪业带来了严重的经济损失[1]。
内质网(Endoplasmic reticulum,ER)是真核细胞内分泌型和跨膜型蛋白合成、折叠、修饰和分选的主要场所,一旦其功能发生紊乱,就会发生内质网应激(ERS),激活由需肌醇酶Ⅰ(Inositol-requiring enzyme I,IRE1)、双链 RNA依赖的蛋白激酶 R样ERS(Protein kinase R-like ER kinase,PERK)和转录活化因子6(Activating transcription factor 6,ATF6)3种内质网跨膜蛋白介导的细胞信号通路来缓和ERS,恢复内质网平衡,这些通路统称为非折叠蛋白反应(Unfolded protein response,UPR)[2]。近年来研究显示,PEDV同别的冠状病毒一样,病毒复制和内质网紧密相关,病毒复制期间合成的大量病毒蛋白在内质网中进行折叠加工修饰,结构蛋白和基因组RNA复制完成后,将在宿主细胞内质网处装配生成新的冠状病毒颗粒,并通过高尔基体分泌至细胞外,这个病毒感染过程常常导致宿主内质网发生混乱,引起ERS[3-4]。葡萄糖调节蛋白78(Glucose regulated protein 78KD,Grp78)是位于内质网中的重要分子伴侣,其上调表达常作为ERS被激活的标志[5]。
为了探究PEDV主要结构蛋白S蛋白对细胞ERS的影响,本研究构建了真核表达质粒pcDNA-S,转染Vero-E6细胞。结果显示 PEDV S蛋白诱导了ERS并主要激活了PERK信号通路,激活的PERK通路反过来抑制了病毒的复制,为阐明S蛋白在PEDV的致病机理中的作用奠定了一定基础,也为抗病毒药物的设计提供了新的思路。
1 材料与方法
1.1 病毒、细胞、菌株及载体PEDV CV777疫苗株(KT323979)、Vero-E6细胞系和 pcDNA3.1(-)载体由本实验保存。感受态细胞E.coliDH5α购自TaKaRa公司。
1.2 主要试剂Primer STAR DNA高保真聚合酶、PrimeScrip Ⅱ 1st strand cDNA Synthese Kit、 DL2000 Marker购自TaKaRa公司;限制性内切酶和T4 DNA连接酶购自NEB公司;质粒提取试剂盒、PCR产物纯化试剂盒和胶回收试剂盒购自AXYGEN公司;病毒RNA提取试剂盒和细胞总RNA提取试剂盒购自QIAGEN公司;蛋白酶和磷酸酶抑制剂、NP-40细胞裂解液、BSA蛋白浓度检测试剂盒购自上海碧云天生物技术公司;Lipofectamine 2000、Alexa Fluor 633山羊抗兔IgG抗体、Alexa Fluor 488山羊抗鼠IgG抗体和Alexa Fluor 546山羊抗鼠IgG抗体购自赛默飞世尔科技有限公司;Tunicamycin(Tu)、Salubrinal(SA)和 DAPI购自 Sigma公司;抗Grp78多克隆抗体、抗p-PERK单克隆抗体(MAb)、抗 p-eIF2α MAb、抗 ATF6多克隆抗体和抗 β-actin MAb购自 Abcam公司;抗 PERK MAb和抗 eIF2α MAb购自Santa Cruz公司;鼠抗PEDV疫苗株CV777 S蛋白MAb和N蛋白MAb由本实验室制备;IRDye800标记的驴抗鼠IgG抗体和IRDye800标记的驴抗兔IgG抗体购自LI-COR公司。
1.3 PEDV感染诱导宿主细胞ERS的检测当Vero-E6细胞长满单层时用1 MOI PEDV感染细胞,分别于感染后6 h、12 h、24 h、36 h和48 h后收集样品,设立Tunicamycin(Tu)阳性对照组和DMSO阴性对照组。分别用荧光定量 PCR[6]和western blot方法检测PEDV ORF3基因和Grp78编码基因mRNA转录水平以及PEDV N蛋白和ERS Grp78蛋白表达水平变化情况。
1.4 重组质粒pcDNA-S的构建与鉴定以PEDV CV777株感染的Vero-E6细胞提取的总RNA为模板反转录制备cDNA,根据GenBank中S全长基因序列(JN599150.1)设计引物,并在其上、下游引物中分别引入KpnⅠ和EcoRⅠ酶切位点(表 1),PCR扩增目的基因,克隆于pcDNA3.1(-)载体中,构建重组真核表达质粒pcDNA-S,通过酶切及测序对重组质粒进行验证。
1.5 PEDV S蛋白表达的western blot鉴定将pcDNA-S转染Vero-E6细胞,同时设pcDNA3.1(-)载体转染对照组。转染48 h后收集细胞,裂解,SDS-PAGE电泳后,以抗 S蛋白 MAb(1∶500)为一抗,IRDye800标记的驴抗鼠IgG抗体(1∶10 000)为二抗进行western blot检测PEDV S蛋白的表达。
1.6 PEDV S蛋白的表达和间接免疫荧光(IFA)鉴定将pcDNA-S转染Vero-E6细胞,同时设pcD-NA3.1(-)载体转染对照组。转染48 h后,用4%的多聚甲醛固定30 min、0.05%TritonX-100通透15 min、5%脱脂乳37℃封闭2 h后,以1∶500稀释的鼠抗S蛋白MAb为一抗,1∶500倍稀释的Alexa Fluor 546山羊抗鼠IgG抗体为二抗,DAPI染色10 min后,以IFA检测PEDV S蛋白的表达。

表1 扩增和检测引物Table 1 Primers used to amplify and detect the target genes
1.7 ERS标志蛋白Grp78的检测将pcDNA-S转染Vero-E6细胞,同时设pcDNA3.1(-)载体对照组、Tunicamycin(Tu)阳性对照组和DMSO阴性对照组。于转染后48 h收集细胞,分别用荧光定量PCR[6]检测Grp78的mRNA水平变化情况和以抗Grp78多克隆抗体(1∶1 000)和抗 β-actin MAb(1∶500)为一抗,IRDye800标记的驴抗鼠 IgG抗体(1∶1 000)或者IRDye800标记的驴抗兔IgG抗体(1∶1 000)为二抗进行western blot检测Grp78蛋白表达水平变化情况。
1.8 S蛋白与 Grp78蛋白的定位分析将 1 μg pcDNA-S转染Vero-E6细胞36 h后,分别孵育鼠抗S蛋白MAb、兔抗 Grp78多克隆抗体,经PBST洗涤后,再分别孵育 1∶500倍稀释的 Alexa Fluor 488山羊抗鼠IgG抗体和1∶500倍稀释的Alexa Fluor 633山羊抗兔 IgG抗体,DAPI染色后使用激光共聚焦显微镜分析S蛋白和内源性Grp78的定位情况。
1.9 S蛋白对UPR信号通路激活的检测将pcDNA-S转染Vero-E6细胞,同时设pcDNA3.1(-)载体对照组、1 μM的 Tunicamycin(Tu)处理的阳性对照组和DMSO处理的阴性对照组。于转染后48 h收集细胞,以抗 PERK MAb(1∶1 000)、 p-PERK MAb(1 ∶1 000)、 eIF2α MAb(1 ∶1 000)、 p-eIF2α MAb(1 ∶500)、 ATF6 MAb(1 ∶1 000)和 抗 β-actin MAb(1∶500)为一抗,IRDye800标记的驴抗鼠 IgG抗体(1∶10 000)或者 IRDye800标记的驴抗兔 IgG抗体(1∶10 000)为二抗进行 western blot检测 PERK和ATF6信号通路的激活情况;同时将细胞提取总mRNA、反转录成cDNA后,通过PCR扩增剪切的与未剪切的XBP1,随后用PstⅠ酶切,将酶切产物进行琼脂糖凝胶电泳检测IRE1信号通路的激活情况。
1.10 Salubrinal(SA)处理后PEDV感染的检测用100 nM的Salubrinal(SA)预处理长成单层的Vero-E6细胞2 h,同时设DMSO处理组为阴性对照组,1 μM的Tunicamycin(Tu)处理组为阳性对照组;随后用MOI=1的PEDV感染,37℃吸附2 h,DMEM洗3遍,分别加入含有上述药物的DMEM维持液,37℃培养36 h,收集细胞,分别用荧光定量PCR[6]和TCID50检测PEDV感染情况。
2 结果
2.1 PEDV感染诱导ERS检测结果用PEDV感染Vero-E6细胞后,分别采用荧光定量PCR和western blot检测 PEDV ORF3基因和 Grp78编码基因mRNA转录水平,以及PEDV N蛋白和ERS标志蛋白Grp78的蛋白表达水平。结果显示,PEDV感染后随着感染时间的推移,PEDV ORF3基因和Grp78编码基因mRNA转录水平,以及PEDV N蛋白和Grp78的蛋白表达水平均明显上调(图1)。结果表明,PEDV感染能够诱导宿主细胞ERS。
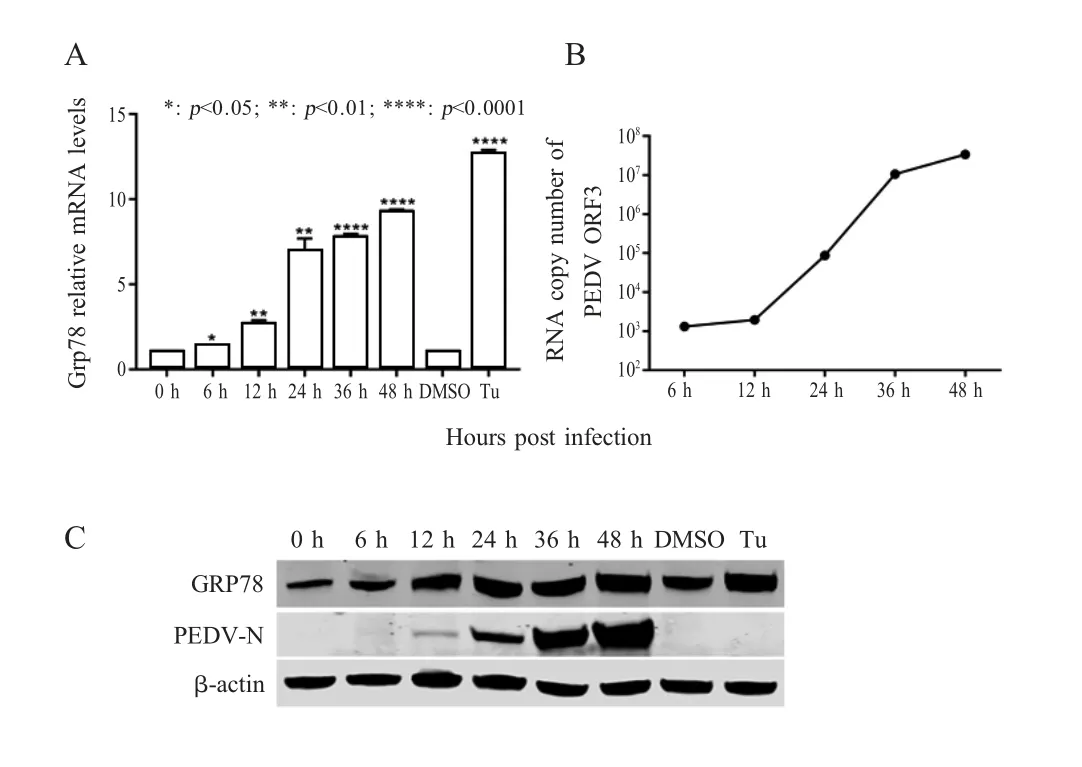
图1 利用 RT-qPCR(A和 B)和 western blot(C)检测PEDV N蛋白和ERS标志蛋白Grp78 mRNA转录水平和蛋白的表达Fig.1 The expression of PEDV N protein and ER stress marker protein Grp78 detected by RT-qPCR(A and B)and western blot(C)
2.2 PEDV S蛋白在Vero-E6细胞内的表达及鉴定将pcDNA-S转染Vero E6细胞,利用western blot检测S蛋白的表达情况,β-actin作为内参对照。结果显示,转染pcDNA-S质粒的细胞在约300 ku处检测到预期大小的条带,而空载体转染对照组在相应位置处没有出现条带(图2A)。同样用S蛋白抗体和Alexa Fluor 546山羊抗鼠IgG抗体通过IFA检测S蛋白的表达情况,同时用DAPI染核后确定S蛋白在细胞内的表达部位。结果显示,转染pcDNA-S质粒的细胞能够观察到红色荧光,而空载体转染对照组未察到红色荧光,S蛋白与DAPI染的细胞核未发生共定位(图2B)。以上结果表明,pcDNA-S质粒能够在Vero-E6细胞的胞浆内表达S蛋白。

图2 PEDV S蛋白表达的western blot(A)和IFA(B)鉴定Fig.2 Identification of the S protein expression of PEDV by western blot(A)and IFA(B)
2.3 PEDV S蛋白诱导了宿主细胞ERS将pcDNA-S质粒转染Vero-E6细胞,用荧光定量PCR和western blot检测ERS标志蛋白Grp78的表达变化,同时用激光共聚焦试验检测S蛋白和内源性Grp78蛋白在细胞内的定位情况。结果显示,pcDNA-S转染组和Tunicamycin(Tu)处理组 Grp78的表达在mRNA转录水平和蛋白水平相对于pcDNA3.1(-)载体转染组和DMSO处理组均明显上调,而外源性表达的S蛋白和内源性的Grp78存在共定位现象(图3)。结果表明,PEDV S蛋白能够诱导细胞发生ERS。

图3 利用RT-qPCR(A)和western blot(C)检测ERS标志蛋白Grp78的表达及用激光共聚焦试验(B)检测S蛋白和内源性Grp78的定位情况Fig.3 The expression of ER stress marker protein Grp78 detected by RT-qPCR(A)and western blot(C)and the colocalization of S protein with endogenous Grp78 identified by confocal images(B)
2.4 PEDV S蛋白对UPR信号通路的影响将pcDNA-S质粒转染Vero-E6细胞,通过western blot检测PERK通路中PERK和eIF2α磷酸化水平来检测PERK通路激活情况;通过western blot检测ATF6蛋白的蛋白酶切割水平来检测ATF6通路的激活情况;通过琼脂糖凝胶电泳检测PstⅠ酶切XBP1后的产物来检测IRE1通路激活情况。结果显示,pcDNA-S转染组和Tunicamycin(Tu)处理组PERK和eIF2α的磷酸化水平相对于pcDNA3.1(-)载体转染组和DMSO处理组明显增加(图 4A和4B);pcDNA-S转染组相对于pcDNA3.1(-)载体转染组ATF6蛋白未发生明显蛋白酶切割,而Tunicamycin(Tu)处理组相对于DMSO处理组发生明显蛋白酶切割现象(图4C);pcDNA-S转染组相对于pcDNA3.1载体转染组XBP1转录本的剪切未发生明显变化,而Tunicamycin(Tu)处理组相对于DMSO处理组发生明显剪切(图 4D);结果表明,PEDV S蛋白主要激活UPR通路中的PERK信号通路。

图4 利用western blot检测PERK通路中PERK与eIF2α的磷酸化水平(A和B)和ATF6通路中ATF6蛋白(C),琼脂糖凝胶电泳检测PstⅠ酶切后的XBP1产物(D)Fig.4 The phosphorylation levels of PERK and eIF2α(A and B)in the PERK pathway and ATF6(C)in the ATF6 pathway detected by western blot,the XBP1 product after PstⅠdigestion detected by agarose gel electrophoresis(D)
2.5 药物Salubrinal增强PERK通路后对PEDV感染的影响检测经典ERS诱导剂Tu处理细胞后对PEDV复制的影响,结果发现Tu处理能够明显抑制PEDV在Vero-E6细胞中的复制,显示ERS抑制PEDV复制。同时使用能够选择性增加PERK信号通路激活的药物Salubrinal(SA)预处理Vero-E6细胞2 h后以1MOI的PEDV感染细胞,36 h后收集细胞,通过荧光定量PCR和TCID50检测PEDV感染情况,确定Salubrinal(SA)增强PERK信号通路后对PEDV复制的影响。结果显示,Salubrinal(SA)处理组能够明显抑制PEDV在Vero-E6细胞中的复制(图5)。表明药物Salubrinal(SA)增强PERK信号通路后抑制了PEDV的复制。

图5 利用 RT-qPCR(A)和 TCID50(B)检测 Salubrinal处理后对PEDV的感染的影响Fig.5 The effect of Salubrinal treatment on PEDV infection detected by RT-qPCR(A)and TCID50(B)
3 讨论
真核细胞内,大多数分泌型和跨模型蛋白的折叠、成熟和修饰发生在内质网腔,在生理状态下,由于细胞分化、增殖和周围环境的变化与细胞的生理状态使得进入内质网的蛋白量会有很大的变化;同样的在一些病理状态下,比如:营养缺乏、缺氧、氧化还原、糖基化和钙离子动员紊乱等状态下均会使内质网的蛋白折叠能力超负荷,引起ERS。为了根据细胞需求快速调整内质网的蛋白折叠能力,细胞激活了称作非折叠蛋白应答(UPR)的信号通路来缓和ERS,恢复内质网的平衡,如果ERS平衡不能够通过自身调节恢复,机体将会诱导凋亡来清除受损细胞。目前已经发现多种病毒感染细胞后同样能够诱导ERS,激活OPR,并最终影响到病毒复制。如非洲猪瘟病毒感染宿主细胞后引起ERS,主要激活UPR信号通路中的ATF6通路促进病毒复制[7];而西尼罗热病毒感染宿主细胞后激活UPR信号通路中的所有3条信号通路来抑制病毒的复制[8]。
最新研究表明,许多冠状病毒感染后可以引起宿主细胞发生 ERS,如 SARS、IBDV等[9-11]。Tong等也报道PEDV感染宿主细胞同样能够引起ERS,并最终抑制病毒的复制[3],与本研究中PEDV感染能够诱导ERS结果一致。也有关于PEDV感染后病毒E蛋白和N蛋白引起ERS,激活UPR的报道[12-14],但对于PEDV最大的结构蛋白S蛋白在病毒感染过程中能否引起ERS,激活UPR却缺乏研究和分析。
因此,本研究构建了PEDV结构蛋白S蛋白的真核表达载体,并展开相关研究。结果显示,S蛋白能够上调细胞中ERS标志蛋白Grp78的表达并且二者在细胞内具有共定位现象,表明S蛋白合成后在内质网内折叠加工修饰,进而积聚诱导了Grp78的上调表达。此外本研究还发现S蛋白主要激活UPR信号通路中的PERK通路,使PERK和 eIF2α的磷酸化水平明显增加,而对ATF6的蛋白酶切割水平和IRE1通路中XBP1转录本的剪切作用却没有明显影响,表明S蛋白没有激活ATF6和IRE1通路,用特异性增强PERK信号通路激活的药物Salubrinal[15]处理能够抑制病毒的复制,提示PEDV感染后由S蛋白诱导的ERS可能通过激活PERK通路的eIF2α的磷酸化降低病毒蛋白合成进而抑制病毒感染,但关于S蛋白能否在PEDV感染的天然宿主细胞-小肠上皮细胞内诱导ERS、激活UPR信号通路以及别的PEDV流行株能否诱导ERS仍然需要进一步的研究。
