Y分子筛中硅铝桥羟基结构调变规律的理论研究
2019-01-17秦玉才宋丽娟
郑 健,李 强,秦玉才,宋丽娟
(辽宁石油化工大学辽宁省石油化工催化科学与技术重点实验室,辽宁抚顺113001)
Y分子筛由于其独特的孔道和酸性质,广泛应用于吸附、分离及催化等领域[1-2]。Y分子筛中为了平衡Al原子的电荷,在其骨架上加入阳离子。HY中的H原子补偿分子筛骨架上的电荷形成桥式羟基,即Brønsted酸(简称B酸)。B酸强度的大小对分子筛催化活性有很大的影响,因此分子筛中B酸酸性的研究一直是大家关注的焦点[3-7]。
对于B酸,其酸强度主要体现的是酸中心提供质子的能力,若O—H键极化较强,羟基质子易动性较大,质子所带的正电荷增加,则B酸强度较强,反之则弱。在HY分子筛中,Si—OH—Al桥羟基作为B酸中心,B酸的强弱对其结构产生影响。而B酸的活性明显依赖于B酸周围的环境。因此,根据化工产品需要,合理调变分子筛的酸性质对研究催化剂的活性有着十分重要的意义。工业中常用的Y分子筛由于合成过程的处理方法,使骨架中的铝原子脱落变成非骨架铝(Extra-Framework Al,EFAl)物种,形成L酸,F.Deng课题组[2,8]通过核磁表征发现,在脱铝的HY分子筛中,EFAl物种的存在能够明显增强邻近B酸的酸强度。
C.J.A.Mota等[9-10]建立6T团簇分子筛模型,对6种单核EFAl物种的落位进行了DFT计算。研究表明,AlO+、Al(OH)2+作为最优的 EFAl物种,可以有效地增强B酸的强度。F.Deng课题组[2,8]结合固体核磁和DFT理论计算研究EFAl物种和B酸活性中心的可接近性,发现在超笼里的Al(OH)3、Al(OH)2+物种和β笼里的Al(OH)2+物种作为最优的L酸活性位存在于脱铝的HY分子筛中。N.Katada等[11]利用周期性密度泛函理论对阳离子改性(Ca2+、Ba2+、Al(OH)2+)的Y型分子筛进行了研究。结果表明,在Y分子筛I'和II'位中,四配位的Al(OH)2+非骨架铝物种是最优的结构,也是提高B酸酸性的关键。随后Pidko课题组[12-13]基于高温条件下非骨架铝物种会自聚形成多核阳离子团簇的理论,建立周期性Y分子筛模型,对各种单双、三和四核EFAl物种的结构及稳定落位进行了相关计算。结果表明,单核四配位的EFAl落在分子筛的SII位上,多核EFAl会进入到β笼,提高距离EFAl物种临近的超笼中的B酸强度。目前有关EFAl物种的结构以及EFAl如何影响B酸性质的研究依然是Y分子筛基础研究的热点。
然而,关于EFAl物种对Y分子筛中B酸周围环境的影响规律依然不明确,B酸强度与EFAl之间的关系是否与他们之间的距离有关,是研究B、L酸协同作用的关键。本文模拟四配位的EFAl物种在周期性的HY分子筛中对B酸结构的影响,利用周期性密度泛函理论计算了HY和FEAl/HY分子筛模型中各个B酸活性中心的性质(结构参数、电子性质、态密度等)。除此之外,还对分子筛B酸中心进行了动力学的计算,探讨B酸强度与EFAl之间的关系,为认识HY分子筛中EFAl与B酸的结构提供理论支持。
1 模型搭建与计算方法
1.1 模型搭建
以往关于Y分子筛的模拟研究主要是基于团簇模型进行的,而本研究模拟了具有Fd-3m对称性、周期性的Y分子筛模型,单位晶胞模型为a=b=c=1.72 nm、α=β= γ=60.0o的三斜菱状模型,骨架中含6个骨架Al原子,所有H原子都置于O1上,此模型记为HY模型。非骨架铝物种选择[AlO]+、[AlOH]2+两种四配位的模型,形成[AlO]+/HY、[Al(OH)]2+/HY周期性模型。
1.2 计算方法
应用美国Accelrys公司的Materials Studio 5.5软件包中的Dmol3模块完成,使用广义梯度近似(GGA)[14-15]中的PBE[16]作为电子交换相关函数,采用可极化的双数值基组(DNP),对于原子中心电子采用DFT semi-core Pseudopotential(DSPP)进行处理。电子自洽计算收敛至1×10-5Ha,为了加速收敛过程,热力学拖尾效应值设为0.002 Ha。
在第一性原理模拟的基础上,利用分子动力学方法研究在常温(298 K)下Y分子筛的稳定性,由此判断B酸位的结构性质。分子动力学采用与第一性原理计算最为相近的Dreiding力场,所有原子的电荷使用第一性原理计算得到的Mulliken电荷,动力学模拟5 ps,每1 fs得到一个构型,用于最后统计分析。
2 结果与讨论
2.1 HY分子筛中B酸活性位结构的解析
对HY分子筛模型进行结构优化,优化的HY分子筛周期性晶胞结构见图1。
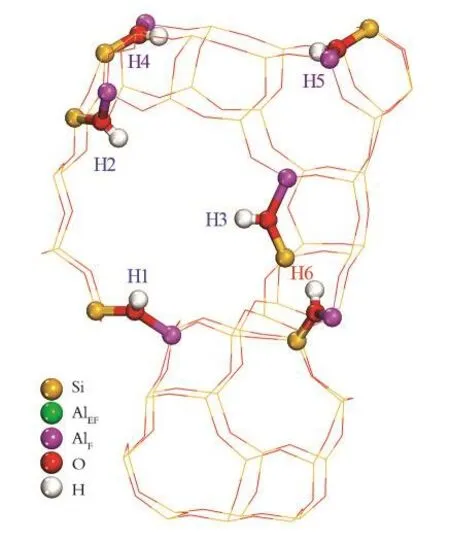
图1 HY分子筛的周期性晶胞结构Fig.1 Periodic cell str uctur e of HY zeolite
由图1可以看出,HY分子筛B酸活性中心的分布是所有H原子均置于分子筛超笼的O1位,其中,H1、H2、H3在超笼的一个十二元环上,H4、H 5在超笼的一个十二元环上,H6在超笼的一个十二元环上。
为了进一步研究这6个B酸活性中心的酸性,对其结构参数进行了分析,表1给出了HY分子筛各个B酸活性中心的几何结构参数。
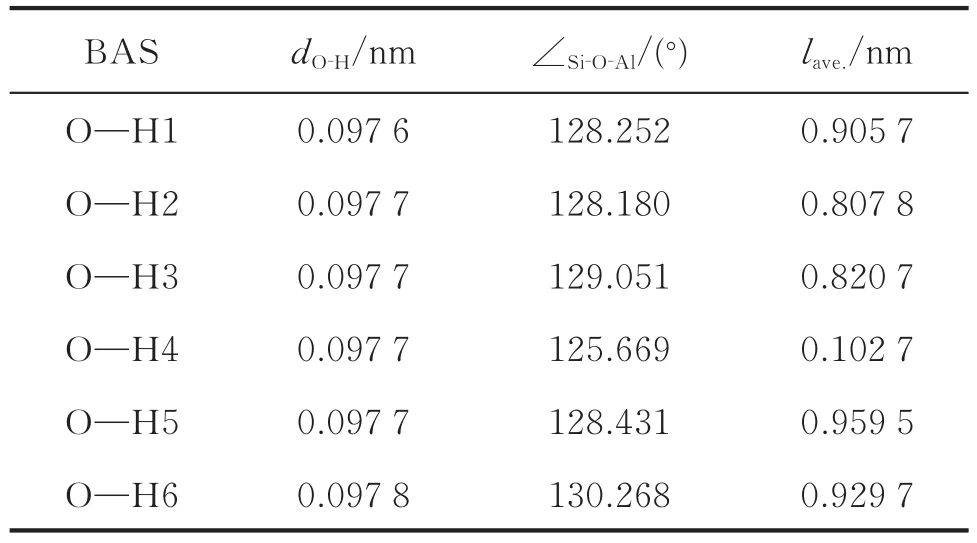
表1 HY分子筛上B酸活性中心的结构参数Table 1 Structural parameters of the B acid site in the HY zeolite
分子筛硅铝桥羟基(Si—OH—Al)上O—H键的键长可以有效地反映B酸位酸性的强弱。键长越长,表明O—H键强度越低,H原子活性越高,酸性质子越容易脱去,固体酸性越强。而硅铝桥羟基上的Al—O键长和Al—O—Si键角参数亦可以反映B酸的强弱[17-18]。这些结论主要基于团簇模型的研究,忽略了分子筛整体对B酸周围环境的影响。由表1可以看出,O—H1和O—H6的键长分别是0.097 6 nm和0.097 8 nm,H 2-H5的O—H键长均为0.097 7 nm。H 6原子所在的十二元环上只有它本身一个B酸活性中心,因此O—H6的键长最长。H 1原子所在的十二元环上除了它本身还有H 2和H 3两个B酸活性位。此外,由表1中lave.数据可以发现,与H2和H3相比,H 1原子与周围B酸中心的平均距离更大,因此O—H 1的距离最小;Si—O4—Al的角度为125.669°,均小于其他的 Si—O—Al的角度;H4与周围B酸活性中心的距离最长,因此Si—O4—Al的角度最小。上述结果表明,B酸结构变化的原因是:分子筛超笼十二元环中B酸个数不同(图1)和B酸周围化学环境不同。因此不能单从硅铝桥羟基结构变化判断B酸强度,需要从电子层面分析B酸的酸强度。
表2给出了HY分子筛上B酸活性中心O和H原子的Mulliken电荷。由表2可以看出,H原子的Mulliken电荷为正值,O原子的Mulliken电荷为负值,这说明H原子与O原子之间发生了电子转移,H原子所带正电荷越多,O原子所带负电荷越少,B酸酸性越强。H原子所带正电荷多少的顺序为H 6>H5>H 4>H2>H 3>H1。其中 H1原子所带正电荷最少,其Mulliken电荷为0.342,H6原子所带正电荷最多,其Mulliken电荷为0.351。结果表明,超笼十二元环上B酸数目越多,其质子H越不易脱去,酸性越弱,即B酸活性中心酸性强弱的顺序为:H6>H 4,H5>H1,H2>H3。O原子所带负电荷多少的顺序为:O5>O6>O1,O4>O3>O2,此顺序与质子H原子的Mulliken电荷的顺序并不对应,导致这种结果的原因是:O原子在骨架上受周围化学环境影响较大,与上述结果一致(见表1)。
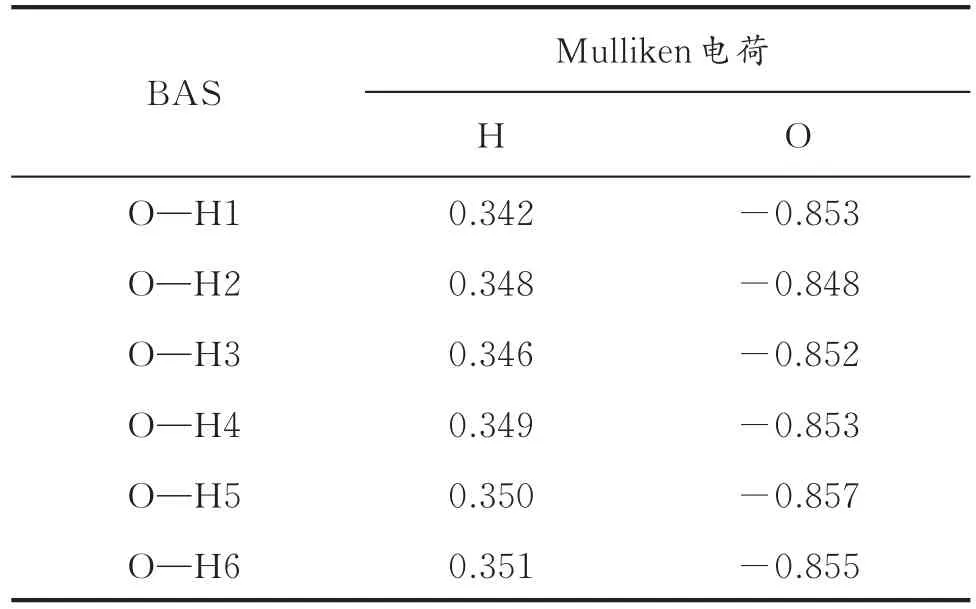
表2 HY分子筛上B酸活性中心O和H原子的Mulliken电荷Table 2 Mulliken charge of O and H atoms on B acid site of HY zeolite
为了进一步研究B酸活性中心的电子性能,计算了HY分子筛B酸活性位质子H的态密度(见图2)。
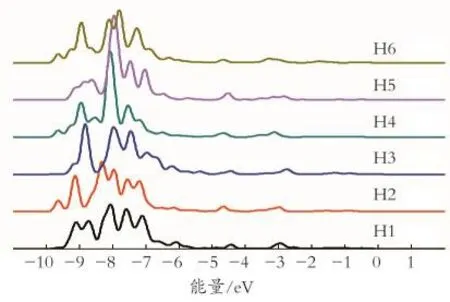
图2 HY分子筛上B酸活性中心H原子的态密度Fig.2 Density of states of H atoms on B acid site of HY zeolite
由图2的H原子态密度曲线中可以看出,H原子的电子主要集中在-6.4~-9.8 eV,根据这区域内的电子分布可以将H原子分为3类:即H 1、H2、H3具有相似的电子分布,H4、H5具有相似的电子分布,H6有它独特的电子分布。这种结果是超笼中H原子个数不同导致的。除此之外,由图2中-2.5~-4.9 eV的电子分布还可以看出,与H 1、H2、H3相比,H 4、H 5、H6的电子局域性更强;还可以发现,H 6在-1~-2 eV有电子分布,电子向费米能级移动,电子变得活跃,因此H 6的酸性最强。即B酸活性中心酸性强弱的顺序为H6>H4,H5>H 1,H 2,H 3,与上述结果相互印证。
2.2 EFAl物种对B酸结构的影响
为了比较不同非骨架铝对HY分子筛B酸位的影响,在HY的分子筛中加入典型的单核四配位EFAl物种([AlO]+,[Al(OH)]2+),对其进行结构优化计算。优化的EFAl/HY分子筛周期性晶胞结构如图3所示。为了研究EFAl对B酸结构的影响,对EFAl/HY分子筛上的质子H进行了重新编号,以下所有H原子的编号都是按照与EFAl的距离长短编排的,如H 1原子就表示与EFAl最近的B酸位H原子。
由图3可以看出,EFAl物种中的Al原子和分子筛SII位的骨架氧配位(O3)形成四配位的稳定结构,[AlO]+物种中的O原子还会和分子筛六元环上的骨架Al成键,使分子筛SII的六元环扭曲变形。
以上结果表明,EFAl的加入引起了骨架活性中心结构的变化,因此对B酸的结构参数进行了分析。表3给出了HY分子筛各个B酸活性中心的几何结构参数。

图3 EFAl/HY分子筛的周期性晶胞结构Fig.3 Periodic cell structure of EFAl/HY zeolite
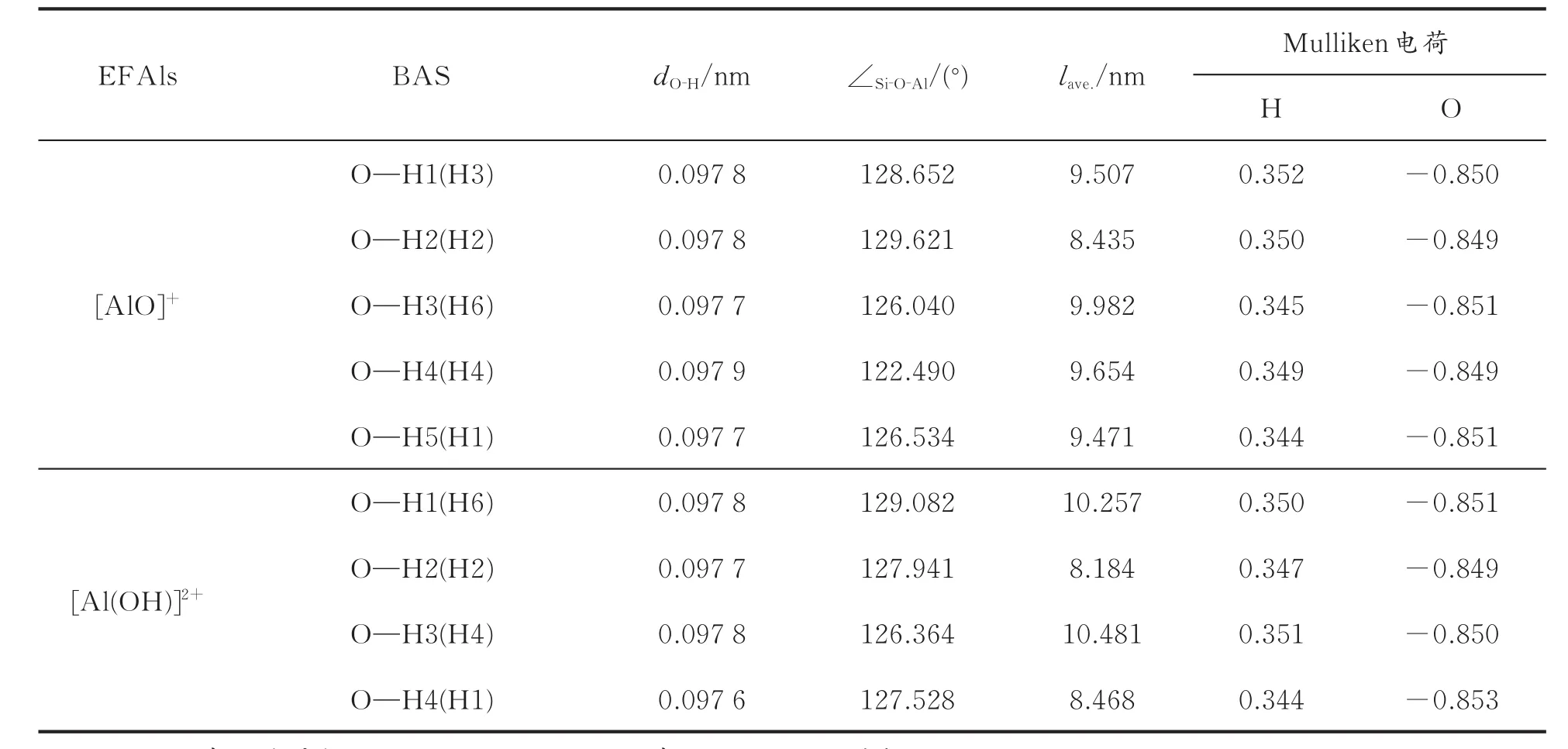
表3 EFAl/HY分子筛上B酸活性中心的结构及电子参数Table 3 Structural and electron parameters of the B acid site in the EFAl/HY zeolite
由表3数据可以看出,部分H原子键长增加了0.000 1~0.000 2 nm,可以认为O—H键长基本没有变化;与O—H键长相比,角度变化比较显著,[AlO]+加入到HY分子筛时,除了Si—O2—Al的角度增加以外,其他角度均变小,通过观察[AlO]+/HY分子筛的结构(见图3)发现,由于[AlO]+中的O与骨架H 1所在的B酸中心的Al原子成键,因此骨架H1和H4所在的十二元环发生移动,使H1和H4的键角显著变小,而H 2原子位于非骨架O原子与骨架Al成键的另一侧,所以只有H2原子使Si—O2—Al的角度变大。当[Al(OH)]2+加入到HY分子筛时,除了Si-O3-Al的角度增加以外,其他角度均变小;同样观察[Al(OH)]2+/HY分子筛的结构(见图3)发现,[Al(OH)]2+物种中O-H的方向偏向H 2的方向,背对H 3的方向,[Al(OH)]2+向H 2的方向偏移,所以只有H3所在的B酸位上Si—O3—Al的角度变大。上述结果表明,EFAl的加入对O—H键长基本没有变化,不同的是Si—O2—Al键角发生了改变,引起角度变化的因素与EFAl物种的形态密切相关。键角的变化不能给出酸性的信息,因此需要从电子层面分析B酸的酸强度,如表3中所示为HY分子筛上B酸活性中心O和H原子的Mulliken电荷。
从表3还可以看出,H原子的Mulliken电荷为正值,O原子的Mulliken电荷为负值,这说明H原子与O原子之间发生了电子转移,而且EFAl的加入增加了部分H原子的Mulliken电荷。影响氢原子电荷变化的因素取决于H原子与EFAl的距离和EFAl的结构。结合表2,计算HY、[Al(OH)]2+/HY和[AlO]+/HY分子筛上B酸位O—H键的平均键长均为0.034 8 nm,所以EFAl的加入对H原子的Mulliken电荷并没有太大的影响。与HY分子筛相比,EFAl/HY分子筛上部分O原子的电子明显减少了,由此可知,EFAl的加入使O原子的电子发生了电荷转移,导致这种结果的原因还与周围化学环境有关,因为O原子在骨架上,受周围化学环境影响较大。
为了进一步研究B酸活性中心的电子性能,分别计算了[AlO]+/HY和[Al(OH)]2+/HY分子筛B酸活性位质子H的态密度(见图4)。
由图4(a)可见,与HY分子筛相比,[AlO]+/HY分子筛上所有H原子的态密度曲线都向左偏移。导致这种结果的原因是:[AlO]+中的O原子与骨架Al成键使分子筛SII位上的六元环发生了变形,引起了H上电子的整体左移。值得注意的是,-3.7~-6.0 eV的电子与HY相比局域性变强,由此可以说明,EFAl的加入增加了B酸的强度。H2-H 5在-7.0~-10.0 eV的电子分布可以看出,H 2和H4的局域性较强。由H1态密度曲线发现,H1中的电子向费米能级移动,电子变得活跃,相比其他H原子,H1原子在-1.0~-1.5 eV和-4.6~-5.5 eV的电子局域性最强。这是由于[AlO]+中的O原子与H 1所在B酸位上的骨架Al原子成键,导致H 1原子比较活泼,因此酸性最强。上述结果说明,EFAl的加入增强了B酸的强度,离EFAl最近的B酸酸性最强。
由图 4(b)可见,能量-3.7~-6.0 eV和-6.5~-9.8 eV的电子局域性比HY和[AlO]+/HY分子筛都强,由此可以说明,EFAl的加入增加了B酸的强度,而且[Al(OH)]2+非骨架物种对B酸的增强比[AlO]+物种要强。从H 2-H 4在-2.5~-4.6 eV的电子分布可以看出,H2的局域性最强,H4的局域性最弱。局域性越强,酸性就越强。由H 1原子的电子态密度曲线可以发现,H1中的电子向费米能级移动,电子变得活跃。相比其他H原子,H 1在-2.5~-4.6 eV和-6.5~-9.8 eV的电子局域性最强,因此酸性最强。上述结果表明,EFAl的加入增强了B酸的强度,B酸活性中心离EFAl越近,酸性越强。
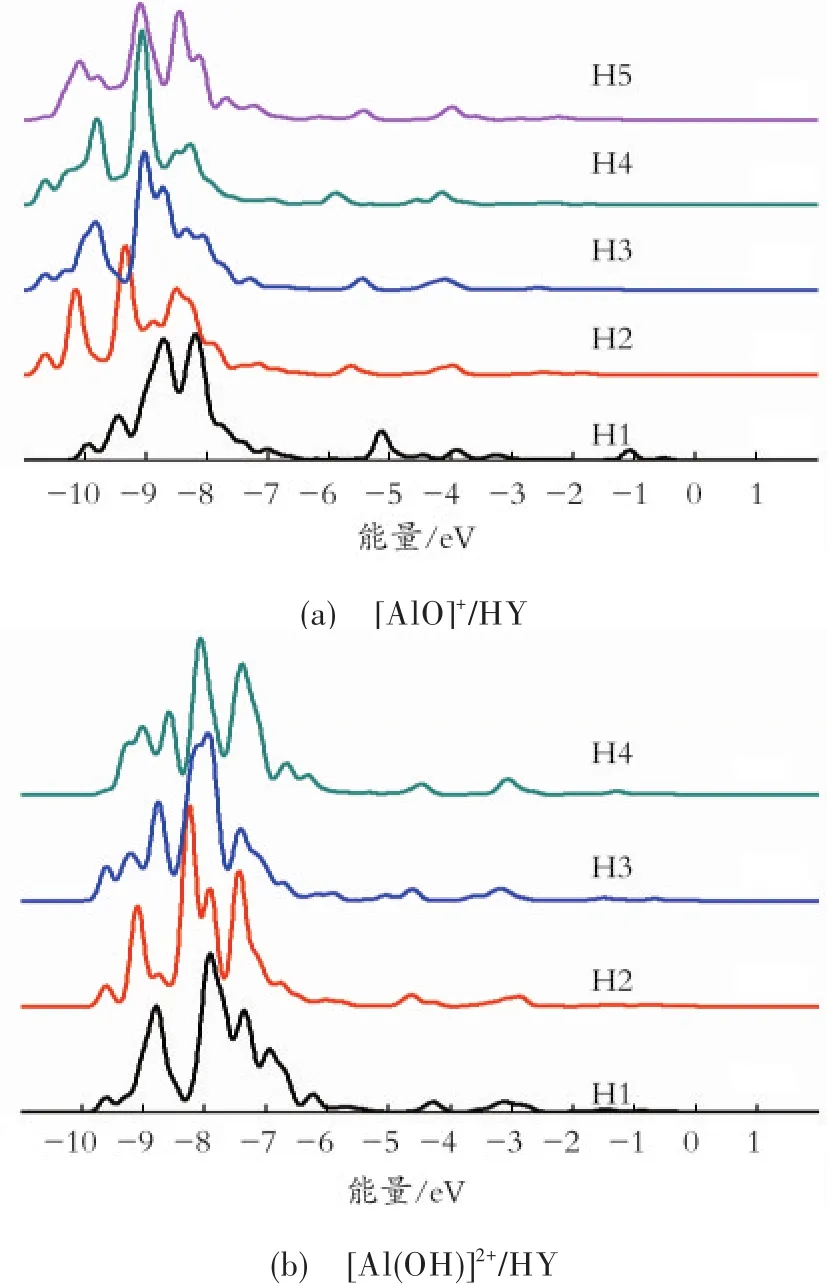
图4 [AlO]+/HY和[Al(OH)]2+/HY分子筛上B酸活性中心H原子的态密度Fig.4 The density of states of H atoms on B acid site of[AlO]+/HY and[Al(OH)]2+/HY zeolite
2.3 分子动力学分析
为了讨论EFAl物种对B酸稳定性的影响,在第一性原理模拟的基础上对周期性模型进行了分子动力学模拟,模拟了室温下(298 K)Y分子筛的动力学性能,计算结果见图5。
由表1可知,HY分子筛H—O键长在0.097 7 nm左右,当EFAl加入后其变化不大,键长只增加了0.000 1 nm。而分子力学的模拟结果显示,H—O键长在0.096 0 nm左右出现的概率最大,这是由于分子力学忽略了电子排斥效应的结果。由图5(b)可知,3种Y分子筛中Si—OH—Al羟基上Al—O—Si键角的分布,通过分子力学计算的键角比量子力学模拟的结果大,说明T原子的位置并不太稳定,这种变化随着非骨架Al的加入而稍有增强,但是整体Y分子筛的结构并没有很大的变化。由图5(c)可以看出,[AlOH]2+/HY模型中H—O键的振动最缓慢,与HY中H原子的运动特性相似,而[AlO]+/HY模型中H原子运动的频率更大,这与实验的结果相呼应。结合图5(a)、(d)也可以看出,[AlOH]2+/HY模型中H、O原子之间的距离分布比另外两个模型更集中于其键长附近,说明H—O键的强度普遍比HY和[AlO]+/HY大,这样的作用更有利于H原子和O原子之间的电荷转移,使B酸酸性变强。
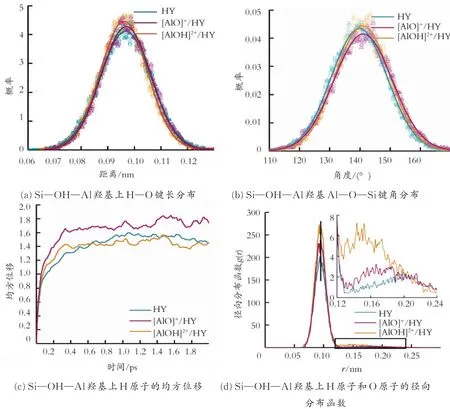
图5 室温下(298 K)Y分子筛的动力学模拟结果Fig.5 Dynamic simulation results for Y zeolite on the room temperature(298 K)
4 结 论
对HY、[Al(OH)]2+/HY及[AlO]+/HY周期性模型进行了第一性原理和分子动力学模拟,研究了B酸中心的结构变化与周围微环境的关系,得到以下结论:
(1)HY中的6个B酸中心中,12元环上H原子的个数与其B酸中心的强度有很大的关系,环上只有1个H原子的酸性最强,含有3个H原子的酸性最弱。在[AlO]+/HY和[Al(OH)]2+/HY分子筛上也有相同的结论。
(2)B酸中心的强度与Si—O—Al的角度关系不大,但是与H—O键长有一定的关系,H—O键长越长,B酸酸性越强。而且在[Al(OH)]2+/HY模型中,H原子振动比较缓慢,这样有利于H原子向骨架移动电子,使H原子的质子性更强。
(3)非骨架铝的加入能增强B酸的酸性,Y分子筛中[Al(OH)]2+物种对B酸的增强量比[AlO]+物种更大。
