Hepatitis C virus core protein modulates several signaling pathways involved in hepatocellular carcinoma
2019-01-16ShahabMahmoudvandSomayehShokriRezaTaherkhaniFatemehFarshadpour
Shahab Mahmoudvand, Somayeh Shokri, Reza Taherkhani, Fatemeh Farshadpour
Abstract Hepatocellular carcinoma (HCC) is the fifth most common cancer, and hepatitis C virus (HCV) infection plays a major role in HCC development. The molecular mechanisms by which HCV infection leads to HCC are varied. HCV core protein is an important risk factor in HCV-associated liver pathogenesis and can modulate several signaling pathways involved in cell cycle regulation, cell growth promotion, cell proliferation, apoptosis, oxidative stress and lipid metabolism. The dysregulation of signaling pathways such as transforming growth factor β (TGF-β), vascular endothelial growth factor (VEGF), Wnt/βcatenin (WNT), cyclooxygenase-2 (COX-2) and peroxisome proliferator-activated receptor α (PPARα) by HCV core protein is implicated in the development of HCC. Therefore, it has been suggested that this protein be considered a favorable target for further studies in the development of HCC. In addition, considering the axial role of these signaling pathways in HCC, they are considered druggable targets for cancer therapy. Therefore, using strategies to limit the dysregulation effects of core protein on these signaling pathways seems necessary to prevent HCV-related HCC.
Key words: Hepatitis C virus; Core protein; Transforming growth factor β; Vascular endothelial growth factor; Wnt/β-catenin; Cyclooxygenase-2; Peroxisome proliferatoractivated receptor α; Hepatocellular carcinoma
INTRODUCTION
At the beginning of the new century, it became clear that infectious agents have an undeniable role in the development of some cancers in humans. It is currently estimated that nearly 16%-18% of all human cancers are attributed to oncogenic viruses[1]. To date, several viruses are linked to cancer in humans, including Epstein-Barr virus (EBV), human papillomavirus (HPV), hepatitis B virus (HBV), human T-cell lymphotropic virus (HTLV), Kaposi’s sarcoma herpesvirus (KSHV), Merkel cell polyomavirus (MCV) and hepatitis C virus (HCV)[2]. HCV is classified as a member of the Flaviviridae family and the Hepacivirus genus. HCV primarily affects the liver and causes chronic HCV infection. Chronic HCV infection inevitably causes additional liver damage, such as hepatitis, cirrhosis and hepatocellular carcinoma (HCC)[3].Globally, an estimated 185 million people, equating to about 2.8% of the world population, have been infected with HCV[4]. Although the prevalence of HCV is declining, the burden of HCV-related mortality due to advanced liver disease is on the rise[4,5]. Two major forms of HCV infection are acute and chronic infection. Acute HCV infection can be seen in nearly 20%-25% of infected individuals, and approximately 15% of these acute infections develop recognizable symptomatic disease[6]. Chronic HCV infection develops in 75%-85% of acute HCV infections, and 10%-20% of all cases with chronic HCV infection slowly progress to liver cirrhosis, of which 1%-5% lead to HCC annually[7]. HCC is a significant health burden worldwide,and it is interesting to note that HCC is the fifth common malignant tumor in men(554000 cases) and the ninth common tumor in women (228000 cases). HCC is the second leading cause of cancer deaths worldwide and was responsible for about 746000 deaths in 2012[8]. Interestingly, 27% and 25% of cases with cirrhosis and HCC,respectively, are associated with HCV infection worldwide[9].
Globally, approximately 399000 deaths per year occur due to HCV-related liver diseases. According to the World Health Organization (WHO) treatment guidelines,more than 95% of HCV-infected patients can be cured by antiviral medicines.Therefore, the use of appropriate antiviral therapy can reduce the risk of death from HCC. The current standard of care for patients with HCV infection is therapy with a novel class of direct-acting antivirals (DAAs) in combination with pegylatedinterferon α (Peg-IFNα) plus ribavirin. To date, the sustained virologic response (SVR)is the best indicator of successful therapy for chronic HCV infection. SVR is defined as having no detectable HCV RNA at 12-24 wk after completion of antiviral therapy, and increasing the chances of achieving SVR is the main goal of treatment[10]. In the treatment course of HCV infection, the rate of SVR has improved to over 95%[11].Several studies showed that the risk of HCC is significantly lower in patients who achieved SVR following antiviral therapy compared to untreated patients[12-14].Overall, more studies are needed to determine whether HCC is reduced among hepatitis C patients after achieving SVR. However, the achievement of SVR is important for HCC prevention. There is currently no prophylactic vaccine for HCV;however, research is ongoing to generate an efficient vaccine[15].
HCV is an enveloped positive-stranded RNA virus that exhibits significant variations across the viral genome. Accordingly, HCV is currently classified into seven genotypes and 67 confirmed subtypes[16]. The HCV genome is approximately 9600 nucleotides in length and encodes a single polyprotein of ~3000 amino acids (aa).The polyprotein is cleaved into ten different structural and nonstructural proteins by viral and cellular proteases. Structural proteins, including core, E1, E2 and p7, are located near the 5′ end of the genome, and nonstructural proteins, including NS1,NS2, NS3, NS4A, NS4B, NS5A and NS5B, are located near the 3′ end of the genome[17](Figure 1). These proteins make numerous interactions with host cell factors involved in important activities such as cell cycle regulation, cell proliferation, cell growth promotion, transcriptional regulation, apoptosis, oxidative stress and lipid metabolism[18,19]. Many lines of evidence clearly indicate that HCV proteins such as core, NS3, NS5A and NS5B can modulate several potentially oncogenic pathways.These proteins also potentiate oncogenic transformation through direct and indirect interactions with various transcription factors and their induction[20-24]. The core protein is an important HCV protein and is responsible for packaging viral RNA and virion budding. This protein (191 aa) is organized into three main domains that include an N-terminal two-thirds hydrophilic domain (D1, approximately 120 aa), a C-terminal one-third hydrophobic domain (D2, approximately 50 aa), and approximately the last 20 aa that serves as a signal sequence for targeting E1 (D3)[25](Figure 1). Kunkel et al[26]showed that the residues 76-113 (tryptophan-rich region) are largely solvent exposed, suggesting that it may interact with cellular proteins. It has been shown that core protein has multi-functional activity and can interact with cellular proto-oncogenes and change their expression patterns, thereby leading to hepatocarcinogenesis[27]. Several lines of investigation have demonstrated that core protein plays a pivotal role in the modulation of several key signaling pathways involved in HCC, such as transforming growth factor β (TGF-β), nuclear factor κB(NF-κB), tumor necrosis factor α (TNF-α), cyclooxygenase-2 (COX-2), Wnt/β-catenin(WNT), vascular endothelial growth factor (VEGF), and peroxisome proliferatoractivated receptor α (PPARα)[22,28-32]. The mechanisms by which core protein modulates these signaling pathways are extremely complicated. To prevent HCV-related HCC,the molecular events underlying the interactions between HCV core protein and the signaling pathways need to be well understood. In this review, we investigate how the interaction of HCV core protein with several signaling pathways contributes to the development of HCC in HCV-infected patients.
TGF-BETA SIGNALING PATHWAY
TGF-β is a multifunctional profibrotic cytokine that is found in three isoforms (TGF-β1-3). Of them, TGF-β1 plays a key role in the pathogenesis of liver inflammation,fibrosis, cirrhosis and HCC[33]. It is interesting to note that TGF-β is considered a central mediator of fibrogenesis and plays an important role in the regulation of tumorigenesis, as it controls numerous cellular functions, including apoptosis,differentiation, proliferation, extracellular matrix production, embryonic development, epithelial-mesenchymal transition (commonly known as EMT), and immune response[34,35]. Fibrosis is one of the most important consequences of TGF-β dysregulation, which is characterized by excessive accumulation of extracellular matrix (commonly known as ECM). TGF-β activity is mediated through activation and proliferation of hepatic stellate cells and connective tissue growth factor.Eventually, progressive fibrosis leads to the development of cirrhosis and HCC[36].TGF-β acts as a double-edged sword depending on the cellular context; in the early stages of cancer development, it exhibits anti-tumor effects, while in the late stages, it has tumor-promoting activities[37]. TGF-β is implicated in several human diseases such as cardiovascular diseases, connective tissue diseases, skeletal and muscular disorders, reproductive disorders, autoimmune disorders, fibrotic disease,atherosclerosis and carcinogenesis[38,39]. Studies have shown that the downstream signaling pathways for TGF-β involve both canonical (Smad-dependent) and noncanonical (Smad-independent) pathways. TGF-β can induce fibrosis via activation of these two pathways, which results in activation of myofibroblasts and excessive production of ECM[40-43]. Smads mediate intracellular responses to TGF-β and have three classes, including receptor-regulated Smads (named R-Smads, including Smad1,2, 3 ,5 and 8), comediator Smads (named Co-Smad, including Smad4) and inhibitory Smads (named I-Smads, including Smad6 and 7)[44]. Smads are regulated via direct phosphorylation by kinase activities of TGF-β receptors (TβRI and TβRII). The model of TGF-β-induced Smad activation is as follows: TGF-β binds to its receptor TβRII,which further interacts and activates TβRI. Activated TβRI then activates Smad2 and Smad3 via phosphorylation. Subsequently, the activated Smad2/3 forms a heterotrimer with Smad4 that translocates into the cell nucleus. Ultimately, the complex associates with other transcription factors and regulates the expression of target genes by binding to promoters containing the minimal Smad binding element[45,46]. Furthermore, TGF-β can activate non-canonical pathways such as JAK,Erk, JNK, p38 MAPK kinase, Ras and RhoA[40,47](Figure 2). It has also been shown that HCV-induced transcription factors such as AP-1, Sp1, NF-κB, EGR-1, USF and STAT-3 can active the TGF-β1 promoter[48]. Taken together, it should be noted that the dysregulation of TGF-β signaling pathway could result in cancer development through either direct or indirect effects on other intracellular signaling pathways involved in carcinogenesis.

Figure 1 Genome organization of HCV. Scheme of HCV core protein domains and functional residues. HCV: Hepatitis C virus.
It has been indicated that oncogenic viruses such as HCV, HBV, HPV, EBV, KSHV and HTLV-1 can modulate TGF-β signaling pathway through various direct or indirect mechanisms, suggesting that this pathway is a desirable target for connecting viral proteins[37]. Several studies support the contention that HCV induces TGF-β1 secretion in HCV patients, as TGF-β1 levels are extremely high in these patients[49-51].The results of the study performed by Jee et al[52]indicated that TGF-β1 protein in HCV-infected cells and in neighboring cells was more than 20-fold higher than in uninfected cells, and this increased production was observed 2 d after HCV infection.Several studies have demonstrated that HCV core protein can induce TGF-β1 promoter activity and directly and indirectly upregulate its gene expression. These findings suggest that HCV infection is one mechanism by which liver fibrosis can be exacerbated[28,53-56]. Taniguchi et al[28]showed that TGF-β1 mRNA expression is increased by HCV core protein, whereas TGF-β2 and TGF-β3 mRNA levels do not change upon core protein expression. The results of their study suggested that bases 331 to 376 in the TGF-β1 promoter are upregulated by HCV core protein[28]. In studies conducted by Battaglia et al[57]and Pavio et al[58], it was revealed that core protein is able to switch TGF-β from a tumor suppressor to tumor promoter by decreasing hepatocyte apoptosis and increasing EMT by decreasing Smad3 activation. The Smad proteins consist of two principal domains, DNA-binding domain (N-terminal Mad homology 1, MH1 domain) and protein-protein interacting module (C-terminal Mad homology 2, MH2 domain). Pavio et al[58,59]indicated that the central domain (59-126 aa) of core protein binds to the MH1 domain of Smad3, leading to TGF-β inhibition.Cheng et al[60]explained that HCV core protein utilizes various mechanisms to regulate TGF-β, including the following: (1) suppression of TGF-β/Smad3-mediated transcriptional activation through interference with the DNA-binding ability of Smad3; (2) block of TGF-β-induced G1 phase arrest via downregulation of TGF-βinduced p21 promoter activation; and (3) resistance to TGF-β/Smad3-mediated apoptosis. Notably, over-expression of HCV core protein indirectly induces TGF-β production by increased reactive oxygen species production, which in turn increases JNK, Erk and p38 MAP kinase activity in a NF-κB-dependent manner[56,61,62](Figure 2).In addition, Shin et al[54]revealed that HCV core protein can regulate other factors associated with fibrosis, such as connective tissue growth factor, TβRII and TGF-β1.Nevertheless, considering the pivotal role of TGF-β in the development of fibrosis and tumor progression, this pathway is an important pharmaceutical target to prevent cancer progression. Furthermore, treatment with antiviral drugs such as DAAs in combination with Peg-IFNα and ribavirin increases the chances of achieving SVR. On the other hand, there are some but not conclusive data that TGF-β1 serum levels in chronic hepatitis C patients under antiviral therapy significantly decreases, especially in patients achieving SVR[63-66]. In this condition, HCV proteins are unable to upregulate TGF-β1 expression, which in turn leads to reduced fibrogenesis and prevention of cancer progression.
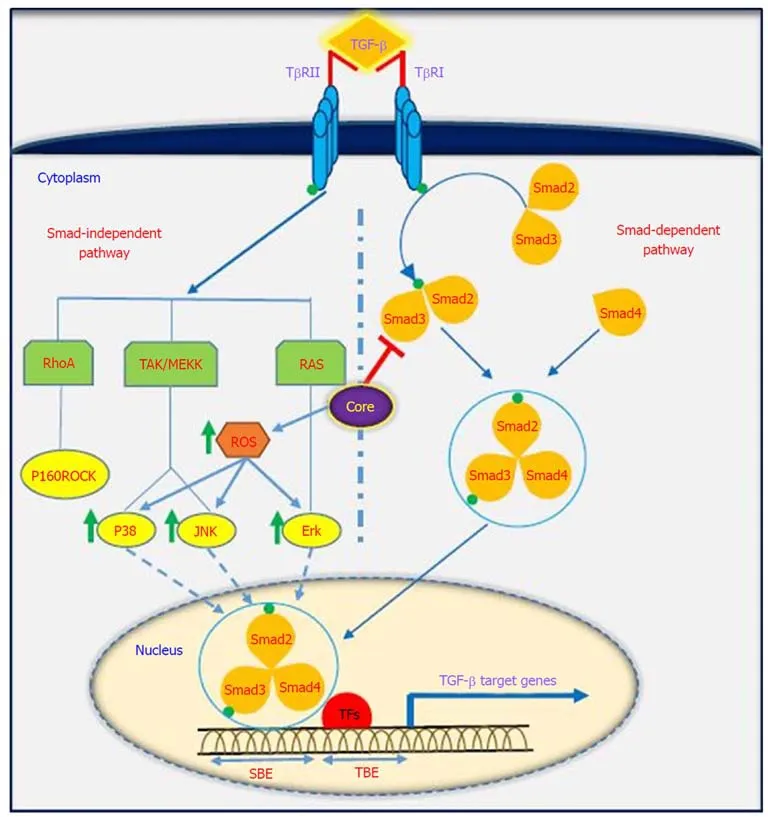
Figure 2 Interaction between the TGF-β signaling pathway and HCV core protein. TGF-β regulates target gene transcription via canonical (Smad-dependent)and non-canonical (Smad-independent) pathways, and HCV core protein modulates TGF-β signaling pathway in various ways. For detailed information, see text. TGF-β: Transforming growth factor β; HCV: Hepatitis C virus; TβR: TGF-β receptor; ROS: Receptor tyrosine kinase c-ros oncogene 1; TFs: Transcription factors; Smad:Small mothers against decapentaplegic; RhoA: Ras homolog gene family member A; MEKK: Mitogen-activated protein kinase kinase; RAS: Rat sarcoma; ERK:Extracellular signal-regulated kinase; JNKs: Jun N-terminal kinases; P160ROCK: Rho-associated coiled-coil containing protein kinase 1; SBEs: SMAD-binding elements; TBE: T-box binding element.
VEGF SIGNALING PATHWAY
VEGF is a signal protein that is produced by most parenchymal cells and stimulates angiogenesis. In cancer progression, VEGF is the key mediator of angiogenesis and vasculogenesis, which leads to the formation of new blood vessels from pre-existing vessels. In turn, this allows tumors to access oxygen and nutrients[67]. VEGFA, also known as VEGF, is a protein with vascular permeability activity that is a member of a family of growth factors. In addition, VEGFB, VEGFC, VEGFD and placental growth factor are also in this family. These growth factors play an important role in angiogenesis and differ in their biological functions and expression patterns[68].Several factors play roles in the upregulation of VEGF. It has been demonstrated that a number of growth factors such as PDGF, epidermal growth factor, fibroblast growth factor, TNF, TGF-β and interleukin-1 can induce VEGF gene expression[69]. VEGF applies its effects by binding to VEGF receptors, which are expressed on vascular endothelial cells. The VEGF receptors include VEGF receptor-1 (Flt-1), VEGF receptor-2 (KDR) and VEGF receptor-3 (Flt-4)[70]. VEGF has several roles, including the following: (1) inducing angiogenesis through a direct impact on endothelial cells; (2)inducing cells to invade the underlying matrix and to form capillary-like tubules; (3)elicitation of non-mitogenic responses by vascular endothelial cells; (4) instrumental in maintaining the viability of immature vasculature and inducing hemotaxis; and (5)the expression of plasminogen activators and collagenases in endothelial cells[71]. It should be noted that the induction and activation of hypoxia-inducible factor-1 alpha(HIF-1α) is a major inducer of VEGF expression in tumors. It is one of the first transcription factors in response to hypoxia and is a closely related angiogenesis factor. HIF-1α responds to hypoxia through binding to hypoxic response element,which in turn leads to an increase in VEGF protein expression because it plays a regulatory role in VEGF expression[72,73]. Given the importance of the angiogenic effects of VEGF, dysregulation of its expression is important in disease processes and progression toward cancer. A large body of evidence exists that suggests a role for VEGF in tumorigenesis in human cancers[74].
Oncogenic viruses such as HCV, EBV, HPV, KSHV and HBV can upregulate VEGF with the use of cellular signaling machinery, which leads to angiogenesis[75]. Several reports have shown upregulation of VEGF in patients with HCV-related HCC[76-80].Mukozu et al[77]demonstrated that VEGF serum levels of patients with HCV-related HCC are significantly higher than those in the control group. HCV core protein can upregulate cellular VEGF expression through HIF-1α transcription factors and activator protein 1 (AP-1). Core protein induces over-expression and stabilization of HIF-1α, which in turn induces VEGF expression[81-84]. Abe et al[82]showed that core protein increases HIF-1α expression level by activating the NF-κB signaling pathway,which leads to an increase in VEGF expression under hypoxia followed by HIF-1α upregulation. They also observed that when cells were incubated with HIF-1α inhibitor, VEGF expression clearly decreased[82]. In a study performed by Zhu et al[83]in Huh7.5.1 cells, it was demonstrated that core protein contributes to VEGF biosynthesis by inducing VEGF expression and secretion. They indicated that using HIF-1α siRNA in Huh7.5.1 cells, which results in reducing expression of HIF-1α,significantly reduces VEGF expression. Shao et al[85]indicated that VEGF expression increased due to activation of AP-1 transcription factor, because the promoter region of VEGF contains binding sites for AP-1 transcription factors, which lead to enhanced VEGF expression via promoter binding. This observation is in line with previous studies of AP-1 activity associated with VEGF expression and HCV core proteininduced AP-1 activity[86-89]. HCV core protein affects androgen receptor (AR)transcriptional activity by activating several signaling pathways such as phosphatidylinositol 3-kinase (PI3K)/AKT and JAK/STAT3. Since VEGF is a target gene for AR in the liver, HCV core protein increases VEGF expression through increased activity of the AR signaling pathway[90]. Hassan et al[31]recently reported that HCV core protein induces VEGF expression mediated by JNK, p38 and ERK signaling pathways (Figure 3). Given the results of various studies, HCV core protein can upregulate the VEGF signaling pathway. These data increase our insights into the molecular mechanisms by which HCV core protein mediates angiogenesis in HCV-infected patients.
WNT SIGNALING PATHWAY
The WNT signaling pathways are a group of signal transduction pathways that regulate different cellular processes such as cell polarity, organogenesis, cell migration and neural patterning during embryonic development[91]. Alteration of WNT activity has been linked to the development of HCC and other liver diseases[92].β-catenin has a crucial role in Wnt signaling and also tightly binds to the cytoplasmic domain of type I cadherins and is implicated in the structural organization and function of cadherins[93]. Wnts are secreted cysteine-rich lipid-modified glycoproteins[94,95]that bind to the N-terminal extra-cellular cysteine-rich domain of the Frizzled (Fz or Fzd) receptor family and low-density-lipoprotein-related protein 5/6 (LRP5/6) as co-receptors[96,97]. When WNT signaling is inactive, cytoplasmic βcatenin interacts with a multiprotein degradation complex comprised of casein kinase I (CK1), adenomatous polyposis coli gene product (APC), glycogen synthase kinase 3β(GSK3β) and Axin[98]. Axin binds to newly synthesized β-catenin, which is subsequently phosphorylated by CKI and GSK3 on conserved Ser and Thr residues in the amino terminus[99]. Following phosphorylation, β-catenin is targeted for proteasome-dependent degradation, including an interaction with β-transducin repeat-containing protein (β-TrCP), a part of the E3 ubiquitin ligase complex, leading to β-catenin ubiquitination and degradation[100,101]. WNT signaling is regulated by secreted proteins, including secreted Frizzled-related proteins (sFRPs) and Wnt inhibitory protein (WIF) that can bind to Wnts and prevent interactions between Wnt and Wnt receptors. Other Wnt inhibitors include Dickkopf (DKK) proteins, which inhibit Wnt signaling by binding to LRP5/6[95]. If the concentration of Wnts increases,Wnts interact with receptors to activate Dishevelled (Dsh or Dvl) protein. Dvl, a modular phosphoprotein, is phosphorylated by several kinases such as CK1.Activated Dvl recruits the destruction complex to the plasma membrane and binds to Axin and Fz[102,103]. Intriguingly, Axin binds to the cytoplasmic domain of LRP5/6.When Dvl is activated, it leads to the inhibition of GSK3 activity, which activates a complex series of events that decreases β-catenin phosphorylation and degradation[104]. Therefore, β-catenin accumulates in the nucleus and activates the transcription of target genes through interaction with DNA-bound T-cell factor (TCF)and lymphoid enhancer-binding factor 1 (LEF) family members[105]. WNT signaling has been implicated in the modulation of innate immunity by stimulating invariant natural killer T cell (iNKT) responses and production of chemokine-like chemotactic factor leukocyte cell-derived chemotaxin 2 (LECT2) or by decreasing the release of tumor necrosis factor[106-108]. LECT2 is involved in inflammation, chemotaxis, immunomodulation, cell proliferation and carcinogenesis. In addition, LECT2 signaling induces inflammatory responses by activating the proinflammatory NF-κB pathway[109]. Abnormal Wnt and Fz expression in hepatocytes, stellate and Kupffer cells might play important roles in liver pathobiology[110,111].
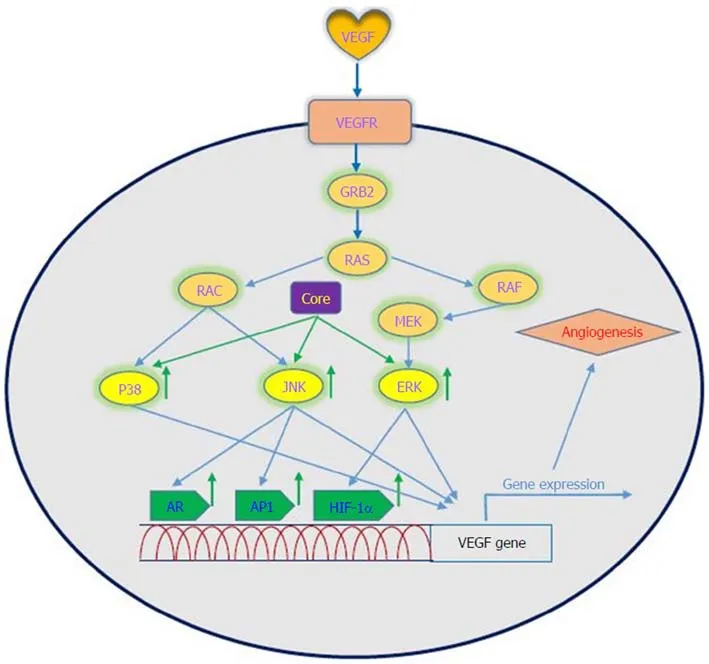
Figure 3 Interaction between VEGF signaling pathway and HCV core protein. HCV core protein upregulates VEGF expression mediated by AR, AP1 and HIF-1α. VEGF: Vascular endothelial growth factor; VEGFRs: Vascular endothelial growth factor receptors; GRB2: Growth factor receptor bound protein 2; RAS: Rat sarcoma; RAF: Rapidly accelerated fibrosarcoma; MEK: Mitogen-activated protein kinase kinase; ERK: Extracellular signal-regulated kinase;JNKs: Jun N-terminal kinases; AR: Androgen receptor; AP1: Activator protein 1; HIF-1α: Hypoxia-inducible factor-1 alpha.
Khanizadeh et al[112]indicated that oncogenic viruses can interact with and modulate the Wnt signaling pathway. Because the WNT signaling pathway is a key pathway in HCV-positive HCC, it is important to understand how HCV core protein induces liver carcinogenesis through Wnt signaling[113](Figure 4). DNA methylation also contributes to the development of HCC. Quan et al[114]showed that HCV core protein can increase Wnt signaling via down-regulation of the sFRP1 promoter in HCC cells through methylation and histone deacetylation. In another study, Umer et al[115]demonstrated that sFRP2 and DKK1 promoters of Wnt inhibitor genes increase DNA methylation in HCC patients compared to normal controls. In addition, HCV core protein is involved in hypermethylation of the CDH1 (E-cadherin) gene promoter. Decreased production of E-cadherin induces Wnt signaling activation[116].HCV core protein enhances β-catenin expression and nuclear stabilization by inactivating GSK-3β. Nuclear accumulation of β-catenin forms a transcriptional complex with TCF and activates downstream target genes, such as c-Myc, Cyclin D1 and WNT1 inducible signaling pathway (WISP-2), which regulate cell growth and cell cycle progression[22,113]. Additionally, HCV core protein elevates the expression of LRP5/6 co-receptors and FZD receptors and releases β-catenin from the β-catenin-E-cadherin complexes[108]. Taken together, the direct involvement of HCV core protein in the Wnt pathway is an attractive candidate to mediate liver pathogenesis.
COX-2 SIGNALING PATHWAY
The cyclooxygenase (COX) family consists of constitutive COX-1, inducible COX-2 and COX-3, which are involved in prostaglandin synthesis by conversion of arachidonic acid to prostanoids, including thromboxanes and prostaglandins[117,118].COX-2 can be induced by tumor promoters, cytokines and growth factors via the cisacting elements within the 5’ UTR of the COX-2 gene and is known as a pathogenic factor involved in cellular proliferation, anti-apoptosis activity, inflammation,fibrogenesis and tumorigenesis. Moreover, increased levels of prostaglandin E2and COX-2 contribute to various biological processes, including oxidative stress, liver damage, bacterial and viral infection, acute and chronic inflammation and cancer[117,119,120].
Studies have shown that there is a close relationship between oncogenic viruses such as HCV, HBV, EBV and HPV and COX-2[121-124]. Previous reports demonstrated that increased production of COX-2 is observed in response to HCV infection[125-127].Overexpression of COX-2 supports HCV replication and has a potential role in hepatocarcinogenesis in HCC and human hepatoma cell lines[121,128,129]. Several studies have previously documented that HCV stimulates COX-2 expression via oxidative stress[130,131]. Oxidative stress is a key contributor in liver fibrosis and carcinogenesis related to HCV infection[132]. HCV core protein upregulates COX-2 levels in hepatocytes and has carcinogenic effects that lead to HCC[62,121,133]. Jahan et al[30]demonstrated that core protein of HCV genotype 3a induces COX-2 expression in Huh-7 cells compared to the core protein of HCV genotype 1a. Conversely, several studies have shown that HCV core protein downregulates COX-2 expression. HCV might avoid the inflammatory responses of host by downregulating this signaling pathway[29,117,134]. COX-2 plays a crucial role in the production of matrix metalloproteinases (MMPs), particularly MMP-2 and MMP-9, by liver cells, which are associated with cell migration via degradation of cellular and extracellular components during organ development, morphogenesis, tissue damage and cancer invasion[135,136]. COX-2 is involved in the regulation of superoxide anion expression,namely NADPH oxidase 1 (NOX1) and NADPH oxidase 4 (NOX4), in hepatocytes[137].Lan et al[138]demonstrated that NOX1 and NOX4 are increased in cirrhotic patients and play an important role in liver fibrosis via regulating proliferation, inflammation and fibrogenesis in hepatic stellate cells. COX-2 stimulates β-catenin activation, which leads to WNT pathway activation and subsequent cell growth and proliferation through its interaction with TCF in the nucleus[139]. COX-2 induces the production of VEGF and the anti-apoptotic proteins of the Bcl-2 family, which are associated with an increase in angiogenesis, invasiveness, and resistance to apoptosis[140]. Leng et al[141]showed a positive correlation between COX-2 and phosphatidylinositol 3-kinase-Protein Kinase B (PI3K-Akt/PKB) pathway activation in human HCC tissue. PI3KAkt/PKB plays a major role in cancer development and progression by inhibiting apoptosis and stimulating cell proliferation. A recent study showed that long noncoding RNA (lncRNA) COX-2 inhibits immune evasion, migration and invasion of HCC cells and may provide a novel theoretical basis for HCC treatment[142]. In view of these facts, there is a direct relationship between HCV core protein and COX-2 overexpression, which in turn leads to alterations in cell signaling pathways (Figure 5). Therefore, the COX-2 signaling pathway should be considered a target to prevent HCV replication and HCV-associated diseases.
PPAR ALPHA SIGNALING PATHWAY
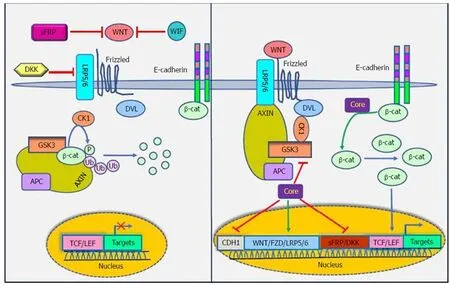
Figure 4 Interaction between Wnt/β-catenin signaling pathway and HCV core protein. HCV core protein increases the expression of Fzd and LRP5/6, decreases the expression of sFRPs and Dickkopf and suppresses the E-cadherin gene promoter. The components shown are explained in more detail in the text. sFRPs:Secreted Frizzled-related proteins; WIF: Wnt inhibitory factor; DKK: Dickkopf WNT signaling pathway inhibitor; LRP5/6: Low-density-lipoprotein-related protein 5/6;Fzd: Frizzled; CDH1: E-cadherin; DVL: Dishevelled segment polarity protein; GSK3: Glycogen synthase kinase 3; CK1: Casein kinase 1; UB: Ubiquitin protein; APC:Adenomatous polyposis coli; TCF/LEF: Transcription factor/lymphoid enhancer-binding factor.
PPARs are a family of nuclear receptor proteins that belong to the steroid/thyroid hormone receptor superfamily and function as transcription factors. This family has three members, including PPARα, PPARγ and PPARβ/δ[143], which play regulatory roles in cellular processes such as differentiation, proliferation, inflammation,oxidative stress, tumorigenesis and metabolism (carbohydrate, lipid, protein)[144]. In order to function, all PPARs initially heterodimerize with the retinoid X receptor(RXR) and then bind to peroxisome proliferator hormone-response elements (PPREs),which are located in the promoter region of PPAR target genes[145]. PPARα is one of the most abundant nuclear receptors expressed in hepatocytes, and it acts as an important and vital regulator in lipid and lipoprotein metabolism. It becomes activated by several molecules such as long-chain unsaturated fatty acids, and once activated, it promotes lipid clearance through β-oxidation upregulation[146]. The relationship between PPARα and cancer development is very complicated.Epidemiological studies have shown that PPARα has carcinogenic consequences in the liver of humans and rodents[147-150]. It is also noted that long-term administration of PPARα ligands can lead to increased ROS generation, accelerated hepatocyte proliferation and development of HCC[151]. In other words, persistent PPARα activation induces hepatic steatosis through increased liver triglyceride synthesis. On the other hand, hepatic steatosis promotes HCC development and acts as an important accelerating factor of HCC in HCV-infected patients[152-154]. Therefore, the hepatic steatosis induced by alteration of fatty acid metabolism in hepatocytes may act as a mediator in causing HCC in HCV-infected patients. Thus, it is important to understand the interaction between HCV infection and PPARα signaling pathway.
On the basis of several lines of evidence, PPARα activity is impaired in patients with chronic hepatitis C infection, which may contribute to hepatocarcinogenesis[32,155].Dharancy et al[32]showed that PPARα expression is significantly decreased in HCV-infected patients compared to the control group. In this study, HCV core protein expression in HepG2 cells led to disruption of PPARα transcriptional activity,demonstrated by decreased expression of the PPARα target gene CPT1A. Shen et al[156]indicated that PPARα has an inhibitory effect on NF-κB. On the other hand, HCV core protein can disrupt PPARα activity. From this point of view, HCV core protein negatively regulates the repressive effect of PPARα on nuclear NF-κB, which in turn leads to NF-κB activation and HCC development. Studies have shown that HCV core protein can indirectly activate PPARα through activation and phosphorylation of ERK1/2 and P38 MAPK[155,157](Figure 6). HCV core protein also binds to RXR and enhances RXR transcriptional activity, leading to the upregulation of some lipid metabolism enzymes. This result suggests that the dysregulation of RXR by HCV core protein may contribute to HCC development[158]. These findings demonstrate that HCV core protein disrupts PPARα activity, leading to HCC in HCV-infected patients.Thus, PPARα can be considered a new therapeutic target for preventing HCC.
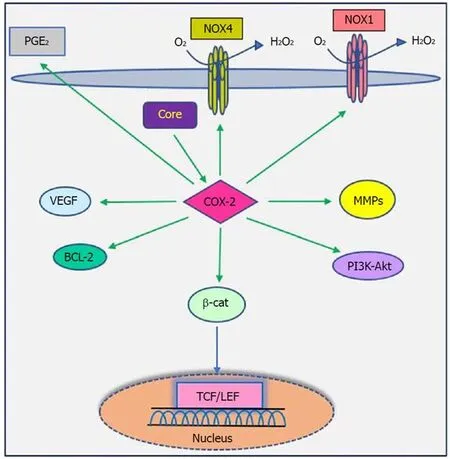
Figure 5 Interaction between COX-2 signaling pathway and HCV core protein. HCV core protein enhances the expression of COX-2 and increases synthesis of PGs, which in turn lead to hepatocarcinogenesis. For detailed information, see text. COX-2: Cyclooxygenase-2; HCV: Hepatitis C virus; PGs: Prostaglandins; PGE2: Prostaglandins E2; NOX: NADPH oxidase; VEGF: Vascular endothelial growth factor; MMPs: Matrix metalloproteinases; BCL-2: B-cell lymphoma 2; PI3K/AKT: Phosphatidylinositide 3-kinases/serine/threonine-protein kinase; TCF/LEF: Transcription factor/lymphoid enhancer-binding factor.
CONCLUSION
HCV can cause liver diseases, especially HCC, through the modulation of various signaling pathways. TGF-β, VEGF, WNT, COX-2 and PPARα signaling pathways play important roles in the regulation of fibrogenesis, angiogenesis and tumorigenesis by controlling cell proliferation, apoptosis, transcriptional regulation and cell growth promotion. Therefore, their dysregulation is associated with HCC. As previously mentioned, HCV core protein uses various mechanisms to dysregulate these pathways. Hence, it seems necessary to undertake major research efforts for a better understanding of how HCV core protein leads to the dysregulation of these signaling pathways, which in turn helps in designing effective therapeutic methods.Furthermore, these signaling pathways should be considered therapeutic targets for cancer therapy, and prevention programs should be implemented to prevent their overexpression. Given the commercially available inhibitors against these pathways,HCV-related HCC development can be prevented in the near future. Moreover, the use of antiviral drugs such as DAAs in combination with Peg-IFNα plus ribavirin leads to SVR in chronic hepatitis C patients, which in turn can help reduce some of the factors involved in cancer development, such as TGF-β1. However, these findings require more research.
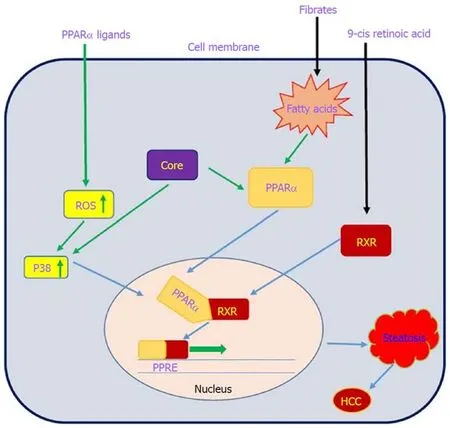
Figure 6 Interaction between the PPARα signaling pathway and HCV core protein. HCV core protein can directly and indirectly activate PPARα, and PPARα activation increases liver triglyceride accumulation, leading to hepatic steatosis. See the text for more details. PPARα: Peroxisome proliferator-activated receptor α;ROS: Receptor tyrosine kinase c-ros oncogene 1; RXR: Retinoid X receptor; PPRE: Peroxisome proliferator hormone response elements.
杂志排行

World Journal of Gastroenterology的其它文章
- Endoscopic foregut surgery and interventions: The future is now.The state-of-the-art and my personal journey
- Role of surveillance imaging and endoscopy in colorectal cancer follow-up: Quality over quantity?
- Initial management for acute lower gastrointestinal bleeding
- Endoscopic trans-esophageal submucosal tunneling surgery: A new therapeutic approach for diseases located around the aorta ventralis
- Autonomic functions and gastric motility in children with functional abdominal pain disorders
- Usefulness of urinary trypsinogen-2 and trypsinogen activation peptide in acute pancreatitis: A multicenter study in Japan