藿香正气丸的质量标准研究
2019-01-15陈雪琴周仔莉贾文江王艳萍
陈雪琴,周仔莉,贾文江,王艳萍
(商洛市药品检验所,商洛 726000)
藿香正气丸(水丸及浓缩丸)是由广藿香、紫苏叶、白芷、白术(炒)、陈皮、半夏(制)、厚朴(姜制)、茯苓、桔梗、甘草、大腹皮、大枣和生姜制成[1],具有解表化湿、理气和中的功效[2-8],用于外感风寒、头痛昏重等症,临床一般用于治疗感冒、荨麻疹、酸中毒和气滞胃痛等病症[9-14]。此方制剂简单、疗效明确,已被应用多年,但现有的质量标准仅限于简单的显微观察及一些常规检查项,为更好地控制其质量,参考对加味藿香正气丸的研究[15-19]及藿香正气水、藿香正气滴丸等的标准[20-26],本文分别对藿香正气丸水丸和浓缩丸主要成分进行TLC鉴别和含量分析研究,以期为其提供更全面的质控标准。
1 仪器与试药
1.1仪器 Waters e2695型高效液相色谱仪(美国沃特世公司);赛多利斯SQP型电子分析天平(北京赛多利斯科学仪器有限公司);AS1512560型超声波清洗机(天津奥特赛恩斯仪器有限公司);四号药筛(湖南省常德粒度分析仪器厂)。
1.2试药 橙皮苷对照品(批号110721-201617,质量分数为96.1%),和厚朴酚对照品(批号110730-201614,质量分数为99.3%)以及厚朴酚对照品(批号110729-200411)均购自中国食品药品检定研究院;藿香正气丸(水丸)(批号160501,陕西香菊药业集团有限公司),(批号:007509,007607,陕西汉王药业有限公司);藿香正气丸(浓缩丸)(批号160504,南京同仁堂药业有限责任公司),(批号20160201,黑龙江葵花药业股份有限公司),(批号20150807,马鞍山天福康药业有限公司),(批号14C30,兰州佛慈制药股份有限公司);乙腈、甲醇均为色谱纯;水为超纯水。
2 方法与结果
2.1广藿香和厚朴的TLC鉴别 取本品浓缩丸2.5 g(水丸6 g),研细,加乙醚20 mL,超声处理(150 W,45 Hz)15 min,过滤,挥干滤液,残渣加乙酸乙酯1 mL溶解,作为供试品溶液。另取和厚朴酚、厚朴酚及百秋李醇对照品适量,分别加乙酸乙酯制成3种质量浓度均为1 mg·mL-1的对照品溶液。分别吸取上述供试品溶液10 μL,3种对照品各5 μL,点于同一硅胶G板上。展开剂为乙酸乙酯-石油醚(60~90 ℃)-甲酸(15∶85∶2),展开,取出,晾干,喷以50 mL·L-1的香草醛硫酸溶液,加热至斑点显色清晰。结果表明,供试品色谱与对照品色谱相应的位置上,显相同的斑点。见图1A。

图1TLC图
A.广藿香、厚朴:1.厚朴酚对照品;2.和厚朴酚对照品;3.百秋李醇对照品;N1~N4.浓缩丸供试品;S1~S3.水丸供试品。B.陈皮:S1~S3.水丸供试品;N1~N4.浓缩丸供试品;1.陈皮对照药材;2.橙皮苷对照品。C.甘草:1.甘草酸铵对照品;S1~S3.水丸供试品;N1~N4.浓缩丸供试品。D.白术:N1~N4.浓缩丸供试品;S1~S3.水丸供试品;1.白术对照药材。E.白芷:S1~S3.水丸供试品;1.白芷对照药材;2.欧前胡素;3.异欧前胡素;N1~N4.浓缩丸供试品。
Fig.1 TLC chromatograms
A.HerbaPogostemonisandMagnoliaofficinalisCortex:1.magnolol reference substance;2.honokiol reference substance;3.autumn lee alcohol reference substance;N1-N4.concentrated pills sample;S1-S3.water pills sample.B.PericarpiumCitriReticulatae:S1-S3.water pills sample;N1-N4.concentrated pills sample;1.PericarpiumCitriReticulatae contrast medicinal materials;2.aurantiamarin reference substance.C.GlycyrrhizaeRadixetRhizoma:1.monoammonium glycyrrhizinate reference substance;S1-S3.water pills sample;N1-N4.concentrated pills sample.D.RhizomaAtractylodisMacrocephalae:N1-N4.concentrated pills sample;S1-S3.water pills sample;1.RhizomaAtractylodisMacrocephalae contrast medicinal materials.E.AngelicaeDahuricaeRadix:S1-S3.water pills sample;1.AngelicaeDahuricaeRadix contrast medicinal materials;2.imperatorin reference substance; 3.isoimperatorin reference substance;N1-N4.concentrated pills sample.
2.2陈皮的TLC鉴别 取本品浓缩丸2.5 g(水丸6 g),研细,加水20 mL,超声处理(150 W,45 Hz)5 min使溶解,加20 mL环己烷振摇提取,分取水层(环己烷液备用),用乙酸乙酯振摇提取3次,每次20 mL,合并乙酸乙酯液(水层备用),挥干溶剂,加甲醇2 mL使残渣溶解,作为供试品溶液。称取0.5 g陈皮对照药材,加20 mL甲醇,超声处理(150 W,45 Hz)30 min,过滤,挥干溶剂,加甲醇1 mL使残渣溶解,作为对照药材溶液。取橙皮苷对照品,制成甲醇饱和溶液,作为对照品溶液。吸取上述供试品溶液5 μL,对照品溶液和对照药材溶液各2 μL,点于同一硅胶G(用40 mL·L-1醋酸钠溶液制备)薄层板上。展开剂为甲醇-乙酸乙酯-水(17∶100∶10),展开,取出,晾干,喷50 mL·L-1的三氯化铝乙醇溶液,置于紫外光灯365 nm处检视。结果显示,供试品色谱中在与陈皮对照药材色谱和橙皮苷对照品色谱相应的位置上,显相同的荧光斑点。见图1B。
2.3甘草的TLC鉴别 取2.2项下的水层,加正丁醇振摇提取3次,每次10 mL,合并正丁醇液,用水洗涤2次,每次10 mL,弃水层,回收正丁醇液的溶剂至干,加甲醇2 mL使残渣溶解,作为供试品溶液。取适量甘草酸铵对照品,加甲醇制成质量浓度为2 mg·mL-1的溶液,作为对照品溶液。分别吸取上述2种溶液各3 μL,点于同一硅胶GF254板上,展开剂为正丁醇-甲醇-氨溶液(8→10)(10∶3∶4),展开,取出,晾干,置于紫外光灯254 nm处检视。结果显示,供试品色谱中,在与甘草酸铵对照品色谱相应的位置上,显相同的斑点。见图1C。
2.4白术的TLC鉴别 取本品浓缩丸2.5 g(水丸6 g),研细,加20 mL正己烷,超声处理(150 W,45 Hz)15 min,过滤后挥干溶剂,残渣加正己烷1 mL使溶解,作为供试品溶液。称取1 g白术对照药材,同法制成对照药材溶液。分别吸取供试品溶液和对照药材溶液各5 μL,点于同一硅胶G板上。展开剂为乙酸乙酯-石油醚(1∶5)(60~90 ℃),展开,取出,晾干,置于紫外光灯365 nm处检视。结果显示,供试品色谱中在与白术对照药材色谱相应的位置上,显相同的荧光斑点。见图1D。
2.5白芷的TLC鉴别 取2.2项下的环己烷液,低温回收溶剂至干,加1 mL乙酸乙酯溶解残渣,作为供试品溶液。取白芷对照药材0.5 g,加乙醚10 mL,浸渍振摇1 h,过滤后挥干滤液,加乙酸乙酯1 mL使残渣溶解,作为对照药材溶液。取异欧前胡素和欧前胡素对照品各适量,分别加乙酸乙酯制成质量浓度均为1 mg·mL-1的2种对照品溶液。吸取供试品溶液和白芷对照药材溶液各5 μL,对照品溶液各3 μL,点于同一硅胶G板上。展开剂为乙醚-石油醚(2∶3)(60~90 ℃),展开,取出,晾干,置于紫外光灯365 nm处检视。结果显示,供试品色谱中,在与白芷对照药材及异欧前胡素和欧前胡素对照品色谱相应的位置上,显相同的荧光斑点。见图1E。
2.6HPLC测定
2.6.1色谱条件的确定与系统适用性实验 填充剂为十八烷基硅烷键合硅胶,流动相A为乙腈,流动相B为水,梯度洗脱,洗脱程序为:0~30 min为25%→75%A,30~40 min为75%→80%A,40~60 min为80%→25%A;流速为1.0 mL·min-1;波长为283 nm;柱温为25 ℃。按照橙皮苷、厚朴酚与和厚朴酚峰计算理论塔板数应不低于3 000,供试品溶液的制备见2.6.3项下方法,取供试品各20 μL,分别注入液相色谱仪,橙皮苷、厚朴酚与和厚朴酚理论塔板数均大于3 000,分离度大于1.5,符合要求。
2.6.2混合对照品溶液的制备 精密称取对照品橙皮苷、和厚朴酚以及厚朴酚11.80,5.0和8.40 mg,置于同一25 mL量瓶中,加甲醇溶解并定容至刻度,摇匀,作为对照品溶液。制成含橙皮苷、和厚朴酚以及厚朴酚对照品质量浓度分别为0.453,0.199和0.336 μg·mL-1的混合对照品溶液。
2.6.3供试品溶液的制备 精密称取研细的藿香正气丸(水丸)和藿香正气丸(浓缩丸)2.17和0.83 g,分别置于250 mL具塞锥形瓶中,精密加入体积分数为70%的甲醇溶液25 mL,混匀,称定质量,超声30 min,放冷,补足减失的质量,过滤,取续滤液,过0.45 μm微孔滤膜,即得供试品溶液。见图2。
2.6.4线性范围考察 取2.6.2项下制备的混合对照品溶液,分别进样2,5,10,15和20 μL,测定峰面积,以对照品的进样量(μg)为横坐标(x)、峰面积值为纵坐标(y)进行线性回归方程,回归方程为:橙皮苷y1=78 439x1+29 815,r1=0.999 0,和厚朴酚y2=21 084x2+16 510,r2=1.000 0,厚朴酚y3=52 791x3-15 754,r3=0.999 5,结果表明,橙皮苷、和厚朴酚及厚朴酚分别在0.95~9.46,0.32~3.16和0.89~8.90 μg范围内有良好的线性关系。
2.6.5精密度考察 精密量取2.6.2项下制备的混合对照品溶液20 μL,连续进样6次,测定其峰面积,橙皮苷、和厚朴酚及厚朴酚峰面积的RSD值分别为0.51%,0.28%和0.54%,结果表明,仪器精密度良好。
2.6.6重复性实验 取同一批藿香正气丸粉末6份,按照2.6.3项下方法制备供试品溶液,进样20 μL,测定峰面积,分别计算含量,橙皮苷、和厚朴酚及厚朴酚含量的RSD值分别为1.59%,1.74%和1.62%,结果表明,该方法的重复性较好。

图2HPLC图
A.混合对照品溶液;B.浓缩丸供试品溶液;C.水丸供试品溶液;1.橙皮苷;2.和厚朴酚;3.厚朴酚。
Fig.2 HPLC chromatograms
A.mixed reference solution;B.concentrated pills sample solution;C.water pills sample solution;1.aurantiamarin;2.magnolol;3.honokiol.
2.6.7稳定性实验 取2.6.3项下制备的供试品溶液20 μL,分别在0,2,4,8,12,18和24 h进行测定,测定其峰面积,结果橙皮苷、和厚朴酚以及厚朴酚3种成分的RSD值分别为1.52%,1.26%和1.71%,RSD值均小于2.0%,该数据符合实验规定。
2.6.8加样回收率实验 精密称取已知含量的过四号筛的同批次浓缩丸样品粉末0.83 g,水丸2.17 g,各平行做6份,分别加入4.008 1和1.887 9 mg的橙皮苷对照品,1.926 4和1.232 9 mg的和厚朴酚对照品以及3.857 0和2.517 2 mg的厚朴酚对照品,按照2.6.3项下方法制备供试品溶液,进样20 μL,测峰面积,计算回收率及RSD值,数据结果见表1和表2。水丸平均加样回收率分别为100.63%,101.41%和100.17%,RSD值分别为0.94%,1.23%和0.45%;浓缩丸平均加样回收率分别为100.87%,101.30%和100.40%,RSD值分别为1.28%,1.14%和1.22%,实验结果表明,供试品溶液的制备准确性高、数据可靠。
表1水丸橙皮苷、和厚朴酚以及厚朴酚加样回收率实验结果
Tab.1 Results of recovery test of aurantiamarin,magnolol and honokiol in water pills
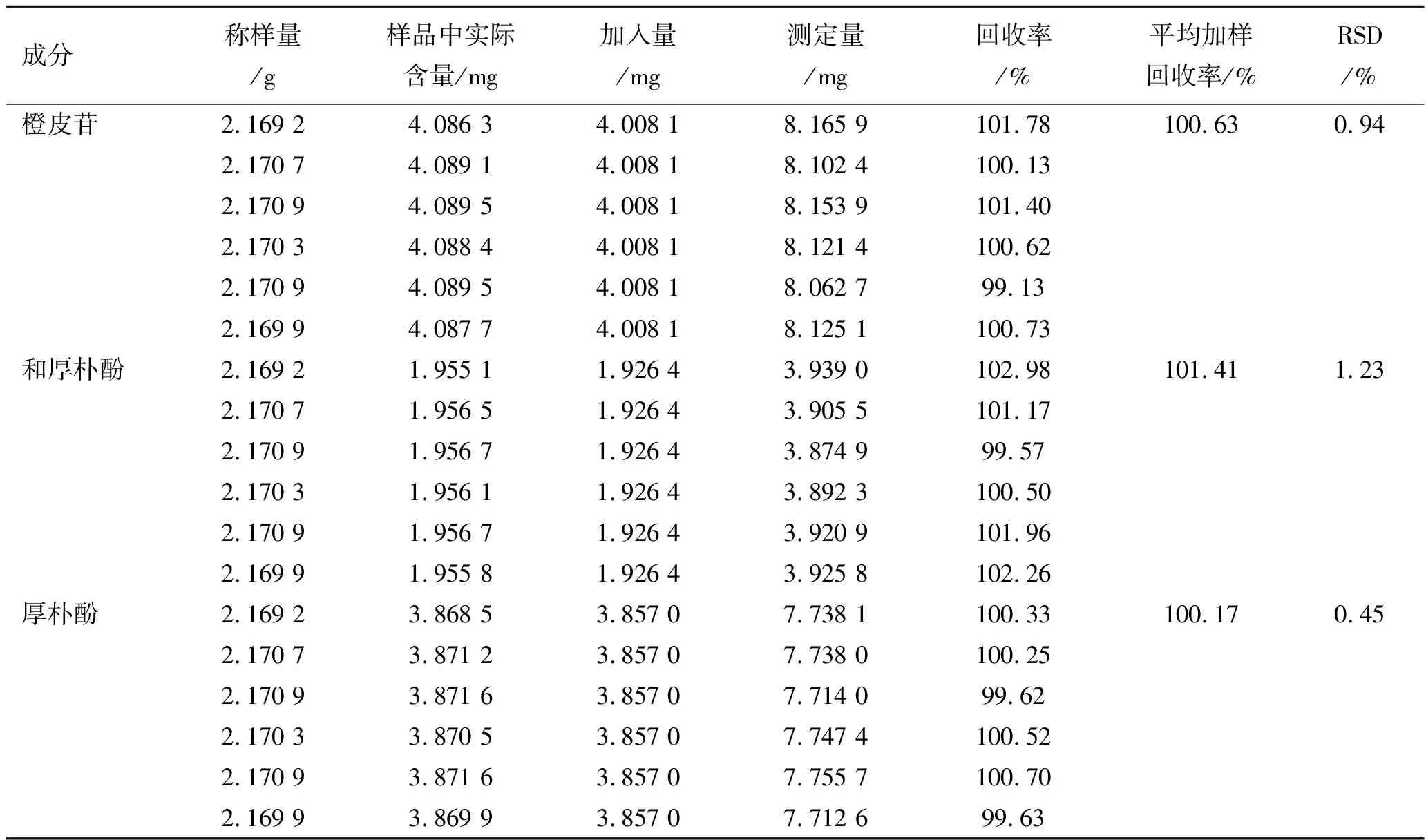
成分称样量/g样品中实际含量/mg加入量/mg测定量/mg回收率/%平均加样回收率/%RSD/%橙皮苷2.169 24.086 34.008 18.165 9101.78100.630.942.170 74.089 14.008 18.102 4100.132.170 94.089 54.008 18.153 9101.402.170 34.088 44.008 18.121 4100.622.170 94.089 54.008 18.062 799.132.169 94.087 74.008 18.125 1100.73和厚朴酚2.169 21.955 11.926 43.939 0102.98101.411.232.170 71.956 51.926 43.905 5101.172.170 91.956 71.926 43.874 999.572.170 31.956 11.926 43.892 3100.502.170 91.956 71.926 43.920 9101.962.169 91.955 81.926 43.925 8102.26厚朴酚2.169 23.868 53.857 07.738 1100.33100.170.452.170 73.871 23.857 07.738 0100.252.170 93.871 63.857 07.714 099.622.170 33.870 53.857 07.747 4100.522.170 93.871 63.857 07.755 7100.702.169 93.869 93.857 07.712 699.63
表2浓缩丸橙皮苷、和厚朴酚以及厚朴酚加样回收率实验结果
Tab.2 Results of recovery test of aurantiamarin,magnolol and honokiol in concentrated pills

成分称样量/g样品中实际含量/mg加入量/mg测定量/mg回收率/%平均加样回收率/%RSD/%橙皮苷0.830 51.885 61.887 93.782 2100.46100.871.280.829 91.884 81.887 93.753 298.970.829 61.884 11.887 93.811 2102.080.829 41.885 41.887 93.821 8102.570.830 81.885 61.887 93.785 3100.630.830 41.885 11.887 93.782 5100.51和厚朴酚0.830 51.247 01.232 92.479 9100.00101.301.140.829 91.246 11.232 92.479 9100.070.829 61.245 71.232 92.498 8101.640.829 41.245 41.232 92.490 7101.000.830 81.247 51.232 92.513 8102.710.830 41.246 91.232 92.509 5102.41厚朴酚0.830 52.510 12.517 25.044 6100.69100.401.220.829 92.508 22.517 24.989 598.570.829 62.507 32.517 25.071 7101.870.829 42.506 72.517 25.009 299.410.830 82.511 02.517 25.063 2101.390.830 42.509 82.517 25.038 9100.48
3 讨论
3.1白术提取试剂的选择 本研究中参考已有文献,选取乙醚和正己烷2种试剂,用同样的方法处理样品,发现对于浓缩丸,2种试剂均可作出理想的TLC图;用乙醚提取的水丸,亦能得到目的斑点,但干扰斑点太多。综合2种丸剂,正己烷更适用。
3.2色谱条件的选择 本文采用乙腈和水梯度洗脱的方法同时测定橙皮苷、和厚朴酚以及厚朴酚的含量,参照采用《中国药典》2015年版藿香正气滴丸、藿香正气水以及藿香正气软胶囊测定橙皮苷、厚朴酚以及和厚朴酚的条件:甲醇-水比例在10%~90%之间等度洗脱测定、甲醇-乙腈-水(40∶20∶40)等度洗脱、乙腈-5 mL·L-1冰醋酸为流动相梯度洗脱、甲醇-水为流动相进行梯度洗脱、乙腈-2 mL·L-1磷酸为流动相进行梯度洗脱等方法,最终经过多次多种方法比较,甲醇-水溶剂系统虽出峰时间快,但橙皮苷分离度和理论塔板数不够、乙腈-5 mL·L-1冰醋酸溶剂系统峰形较差,乙腈-2 mL·L-1磷酸溶剂系统峰分离度不好,峰形较差,最终得到在乙腈-水溶剂系统中橙皮苷及其他2个成分峰的分离度、理论塔板数和拖尾因子均符合要求。经过全波段扫描,结果发现橙皮苷在283 nm波长处有最大吸收,和厚朴酚以及厚朴酚2个成分在293 nm波长处有最大吸收,但三者在283 nm处均能同时出现且都有较强的吸收,方法简便、快捷。
3.3方法选择 本方法借鉴了《中国药典》2015年版藿香正气丸的含量测定方法,对比了橙皮苷、厚朴酚以及和厚朴酚在乙腈、甲醇和水等不同溶剂中的溶解情况,对照品与供试品在甲醇溶剂中的溶解性良好,其液相色谱中色谱峰的保留时间能够保持一致,分离度较高;在15,30,45和60 min的超声时间中,30 min用时最少且效果最好,且超声的方法较萃取、回流等其他方法样品的制备过程简单、高效,最为合理;提取方法中不同比例的甲醇体积分数进行比较之后,浓缩丸和水丸同时在体积分数为70%的甲醇中的溶解性最好,最适宜3个成分的含量测定,且加标回收率较高,说明藿香正气丸3个样品峰在该实验条件下能够被准确定性定量测定,且基线噪声较小,干扰峰较少,准确性、稳定性和重复性都很好,可作为藿香正气丸的质量控制方法。
