pH Eflect on Oxidation of Hydrogen Peroxide on Au(111)Electrode in Alkaline Solutions
2019-01-10JiaojiaoLiJieWeiJunCaiYanxiaChen
Jiao-jiao Li,Jie Wei,Jun Cai,Yan-xia Chen
Hefei National Laboratory for Physical Sciences at the Microscale and Department of Chemical Physics,University of Science and Technology of China,Hefei 230026,China
The hydrogen peroxide oxidation reaction(HPOOR)on Au(111)electrode in alkaline solutions with pH values ranging from 10 to 13 was examined systematically.HPOOR activity increased and the slope of the i-E curve decreased with increasing pH.HO2−is suggested to be the main reactive intermediate for HPOOR in alkaline media.The fast kinetics for HPOOR in alkaline solution is facilitated by the electrostatic interaction between the positively charged electrode and the reactive anions(i.e.,HO2− and OH−),which increases the concentration of these reactants and the thermodynamic driving force for HO2−oxidation at the reaction plane.
Key words: Oxidation of hydrogen peroxide,Au(111)electrode,pH eflect,Electrostatic interaction
I.INTRODUCTION
The oxygen reduction reaction(ORR)is the primary cathodic reaction in fuel cells.Its high overpotential remains the most critical bottleneck restricting the commercialization of fuel cells[1–3].For reactions such as ORR,which involves the transfer of protons and electrons,variation in the solution pH may have many effects on these reactions[2,4,5]:it may change(i)the thermodynamic equilibrium potential,(ii)the nature and concentration of the reactive species,(iii)the reaction kinetics of electrocatalytic reactions,and(iv)the surface properties,e.g.the type and the coverage of the adsorbate and the excess surface charge.Hence,studying the pH eflect will provide rich information towards unravelling the reaction mechanism and identifying the most important factors.
Hydrogen peroxide(HPO)is one of the possible intermediates of ORR[1–3].On Pt group or Pt-based electrocatalysts,there is a potential range in which ORR is under kinetic and mass transport mixed control.In this mixed potential region,both the oxidation and the reduction of HPO(denoted as HPOOR and HPORR hereafter)can occur[1,6].Understanding the mechanism and kinetics of HPOOR and HPORR will be of great help in understanding the mechanism and identifying the key factors governing the efficiency of ORR[2,7–9].However,it remains difficult to obtain a clear understanding of the structure-activity relationship of each reaction on such electrocatalysts.On Au electrodes,a very large overpotential is needed for HPORR to occur[2,9],which leads to a large separation between the potential regions in which HPOOR and HPORR occur.Hence,the Au electrode is a good model system for studying the kinetics and mechanism of HPORR and HPOOR.To the best of our knowledge,only one study has examined HPOOR and HPORR on a Au electrode in acid solution at two diflerent pH values(pH=1.2 and 4.0)[10].This study found that at a constant pH,the HPOOR activity changes with electrode crystal orientation in the order Au(111)
Until now,the pH eflect on HPOOR and HPORR on Au(111)electrodes in alkaline solution has not been reported,in this study,the pH eflect on HPOOR and HPORR on Au(111)in solutions with pH ranging from 10 to 13 is examined.We find that the HPORR activity is negligibly slow on Au(111)in all solutions examined.With the gradual increase of solution pH from 10 to 13,the HPOOR activity increases monotonically.Factors which determine HPOOR and HPORR kinetics on Au(111)will be discussed based on these findings.
II.EXPERIMENTS
Solutionswith 0.5mol/L NaClO4+2mmol/L H2O2+x mmol/L NaOH were prepared using 99%sodium perchlorate(Suprapure,Aladdin),30%H2O2(SSG,Wako Pure Chemical industries,Ltd.),99%sodium hydroxide(Suprapure,Sigma-Aldrich)and Milli-Q water(18.2 MΩ·cm).The working electrode(WE)Au(111)(diameter:2.5 mm)was prepared by the Clavilier method[11].The annealing and cleaning of the WE were carried out in the same procedure as described in Refs.[2,7,9]. A conventional two-compartment,three-electrode glass cell was used for all the electrochemical measurements.An Ag/AgCl electrode and an Au foil electrode were used as reference electrode(RE)and counter electrode(CE),respectively.A meniscus configuration was maintained between the Au(111)surface and the electrolyte during all measurements.The oxidation of H2O2on Au(111)was investigated in N2-saturated(99.999%,the Linde Group,China)solution by cyclic voltammetry using a hanging-meniscus rotating disk electrode(HMRDE)configuration at various electrode rotation speeds and a scan rate of 50 mV/s.The electrode rotation speed(ω)was controlled using a modulated rotator(Hokuto Denko Ltd.).All potentials in this work are quoted against the reversible hydrogen electrode(RHE).
III.RESULTS AND DISCUSSION
A.Characterization of the Au(111)electrode
The quality of the prepared Au(111)electrode was examined by cyclic voltammetry in N2-saturated 0.1 mol/L H2SO4(inset in FIG.1).In the positivegoing scan,small anodic currents at low potentials(<0.2 V)correspond to capacitive charging of the electrical double layer.Scanning of the electrode potential toward more positive potentials leads to the lifting of the thermally induced reconstruction,from Au(111)-to Au(111)-(1×1),which is represented in the voltammogram by the current peak at ca.0.55 V.The broad feature in the potential region from 0.40 V to 0.80 V is attributed to the disordered adsorption of(hydrogen-)sulfate anions on Au(111)-(1×1). A disorder-order phase transition takes place within the sulfate adlayer upon reaching a critical coverage of 0.20 ML,as represented by the pair of sharp current peaks at ca.1.05 V.The current-potential(i-E)features of the Au(111)electrode agree well with those reported in Refs.[12,13],demonstrating the high quality of the Au(111)electrode employed in this study.

FIG.1 Cyclic voltammograms of Au(111)electrode in 0.5 mol/L NaClO4 +x mmol/L NaOH solutions with diflerent pH.Scan rate is 50 mV/s.Inset shows the CV for the same electrode recorded in 0.1 mol/L H2SO4 .
Before investigating the electrochemical behavior of HPOOR,the base cyclic voltammograms were also recorded in alkaline solutions without H2O2,as shown in FIG.1.The small anodic current appearing in the positive-going scan from 0.3 V to 1.0 V comes from the discharge of OH−,which leads to the formation of adsorbed OH(denoted as OHad).At an electrode potential E>1.0 V,surface oxidation of Au(111)takes place,with two anodic peaks appearing at ca.1.1 V and 1.3 V,respectively.Reversing the scan direction at 1.5 V results in a rapid cessation of oxidation currents,followed by a cathodic peak at ca.1.1 V,due to the reduction of three diflerent gold oxides in diflerent oxidation extent(Au+,Au2+and Au3+)[10].These features are quite similar to those reported in acidic media[10,12,13],except that the potential regime for the adsorption/desorption of OH and the oxidation/reduction of Au is negatively-shifted by ca.60 mV compared with that in acidic media.The high OH−concentration of the alkaline solutions used in the present study is suggested to be the origin of these changes.Furthermore,we find that the CV features of Au(111)in solutions with diflerent pHs diflering a little in the potential region with E>1.0 V for Au oxidation(FIG.1(b)).This is probably due to the slight diflerences in the meniscus height,together with the high viscosity of the electrolyte,which leads to the involvement of diflerent numbers of sites(most defects)on the edge in the electrochemical processes.We will demonstrate in a forthcoming work that due to the great diflerence in the HPOOR activity of Au(111)sites and defect sites at the edge,significant diflerences can be seen in the polarization curves for HPOOR.All these remind us that carefully controlling the meniscus height is of great importance in order to remove such eflects when studying the eflect of pH on the HPOOR on Au(111).
B.pH eflect on HPOOR on Au(111)electrode
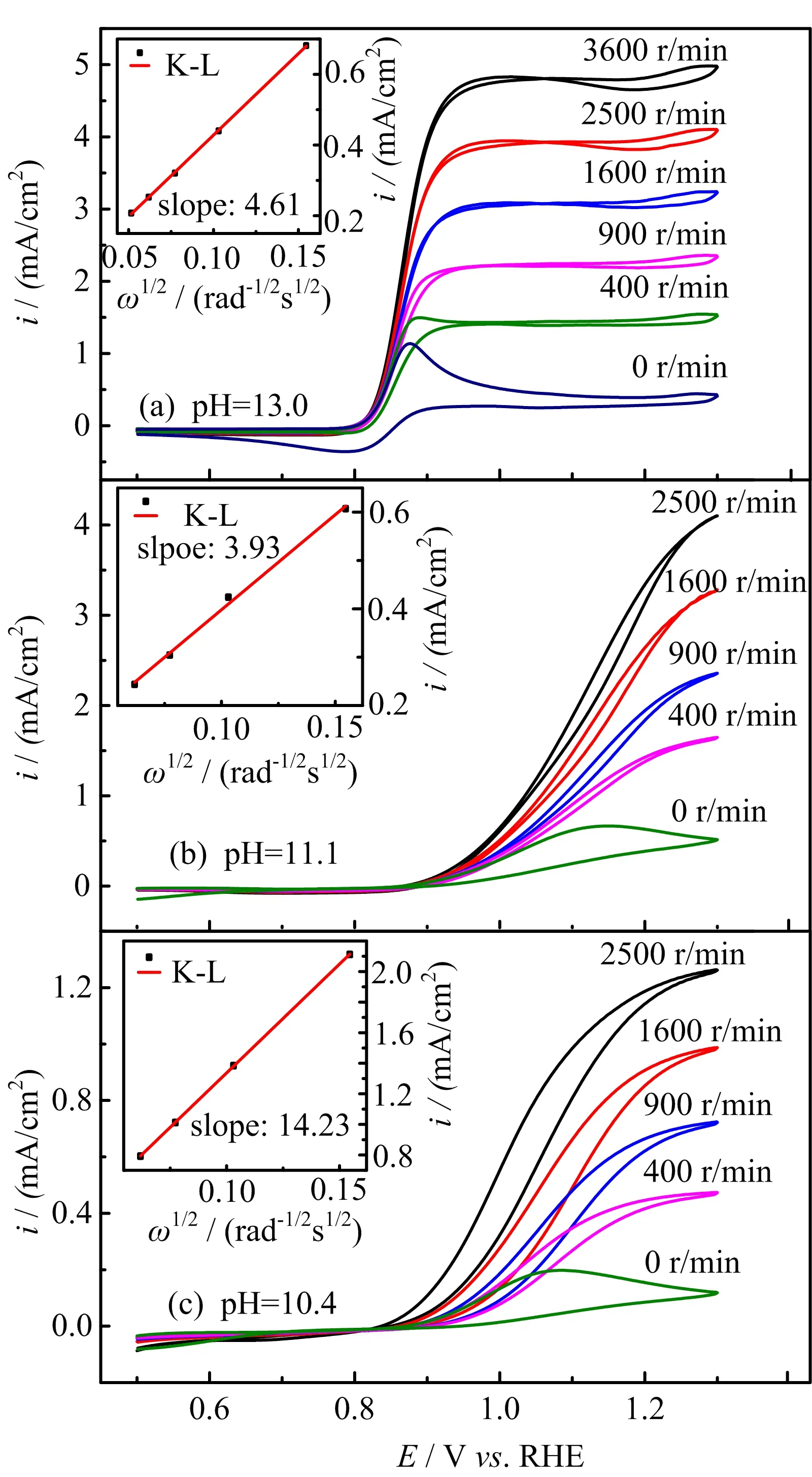
FIG.2 Polarization curves for the oxidation of H2O2/HO2−on Au(111)electrode in 0.5 mol/L NaClO4 +2 mmol/L H2O2+x mmol/L NaOH with pH of(a)13,(b)11.1 and(c)10.4 at various electrode rotation speeds,as indicated in the figure,scan rate:50 mV/s.Inset in(a)shows the diflusion-limited current as a function of the root square of the electrode rotation speed.
FIG.2(a)displaysthepolarization curvesfor HPOOR on a Au(111)electrode in 0.1 mol/L NaOH at diflerent electrode rotation speeds.In the positive-going scan from 0.5 V to 0.8 V,there is barely any current.HPOOR starts at E>0.8 V.The oxidation current increases with E and reaches a plateau at E>0.9 V.The onset potential for HPOOR being ca.0.8 V,ca.0.16 V more positive than its thermodynamic equilibrium potential.The diflusion-limited current for oxidation of H2O2or HO2−is reached because the concentration of hydrogen peroxide is rather low(2 mmol/L).The i-E curve in the potential regime from 0.8 V to 0.9 V is quite close to the one limited by mass transport effect with a Tafel slope near the onset potential below 30 mV/dec.When reversing the potential scan direction at 1.3 V,the HPOOR current in the potential region from 1.3 V to 1.1 V is slightly smaller than that in the positive-going scan,while at lower potentials,the HPOOR current remains nearly the same as that in the positive-going potential scan for the cases with electrode rotation speed(ω)higher than 400 r/min.
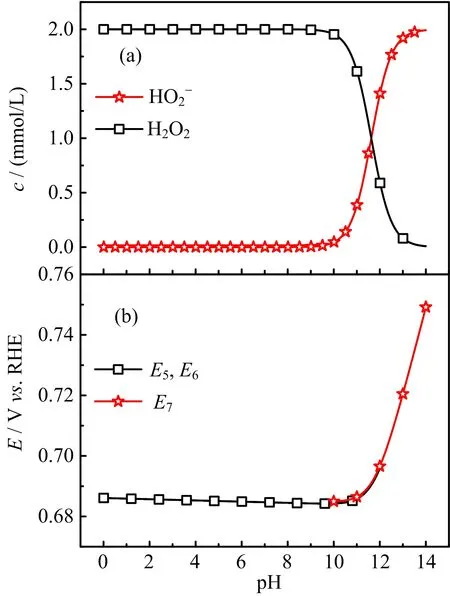
For the cases with ω≤400 r/min,the slight decrease of HPOOR current at E>0.9 V is due to the fact that mass transport rate of H2O2to the electrode surface is slower than the rate of its consumption.As a result,the H2O2concentration as well as its concentration gradient decreases as the potential is made more positive.The inset(FIG.2)shows the diflusion-limited current plotted as a function of the square root of the electrode rotation speed,which displays a well-defined linear behavior.From the slope,2 electrons are estimated to be transferred,confirming the complete oxidation of H2O2on Au(111)electrode.For the cases in which the electrode rotation speed is higher than 400 r/min,barely any cathodic current is observed at 0.5 V The general features for HPOOR on Au(111)in solutions with pH=11.1 and 10.4 are similar to that with pH=13.For all examined pHs,there is barely any current measured for E<0.8 V.At E>0.83 V,a net anodic current flows,which increases with electrode potential and electrode rotation speed.Furthermore,the i-E features in the subsequent,negative-going scan follow roughly that in the positive-going scan in all examined solutions.Besides such similarities,several significant diflerences are noticed:at the same potential,HPOOR current decreases significantly with the decrease of solution pH from 13 to 10.As a result,the slope of the i-E curve decreases,and the potential at which the plateau is reached shifts to higher potential.For the case with pH=10.4,the HPOOR current at 1.3 V is only ca.1.25 mA/cm2,which is ca.1/4 of the diflusion-limited current detected at E>0.95 V in solution with pH=13.The pH dependence of the i-E curves for HPOOR is seen more clearly in FIG.3,where the i-E curves for HPOOR on Au(111)recorded under 2500 r/min in solutions with diflerent pHs are shown.Compared with the case in 0.1 mol/L HClO4,we found that in 0.1 mol/L NaOH the onset potential for HPOOR is ca.0.2 V more negative.With the pH decrease of solution,although the onset potential does not show obvious positive shift,the Tafel slope displays a clear increase,which is more than 120 mV/dec in solution with pH=10.4.Furthermore,we found that in all solutions there is barely any HPORR activity on perfect Au(111)in the potential regime from 0.5 V to 0.8 V.This is probably because the interaction of H2O2(or HO2−)with Au(111)is too weak,since the d-band center of Au(111)is ca.3 eV lower than its Fermi level[3]. FIG.3 Polarization curves for the oxidation of H2O2or HO2−on Au(111)in 0.5 mol/L NaClO4 +2 mmol/L H2O2+x mmol/L NaOH.pH values are indicated in the figure.Electrode rotation speed:2500 r/min,scan rate:50 mV/s. In the following,we will focus on the pH eflect on the HPOOR at a Au(111)electrode in alkaline media.Before discussing the origins of the pH eflect on the HPOOR,we will first give an overview of how the concentrations of the reactants,as well as the equilibrium potential for HPOOR,change with the pH solution.When changing the solution pH,the following reactions will occur The pH-dependences of the H2O2and HO2−concentration are: The pKaof H2O2is 11.8,the pH dependences of HO2−and H2O2concentration in solutions prepared with 2 mmol/L H2O2are plotted in FIG.4(a).From FIG.4(a),we see that when the pH is below 10,H2O2is the main species,while with 10 The equilibrium potentials for reactions(5),(6),and(7)are: FIG.4(a)Concentration and(b)equilibrium potential for the oxidation of H2O2and HO2−as a function of solution pH.The total concentration of H2O2+HO2−is 2 mmol/L. Assuming that the concentration of O2near the electrode surface in all solutions is ca.1.0×10−3mol/L,the pH dependencies of the equilibrium potentials of reactions(5),(6)and(7)can be estimated.The results are plotted in FIG.4(b). When pH is smaller than 11.8,the dominant reactive species in the solution is H2O2.From FIG.4(b),we see that equilibrium potential-pH relations for reactions(5)and(6)are identical.On the RHE scale it does not change with solution pH in the range from 0 to 12.For reaction(7)with HO2−and OH−as reactants,its equilibrium potential increases with increasing solution pH.In the solutions with pH=10 and 11.8,the Eeqfor reaction(7)are ca.0.17 V and 0.06 V more negative than those for reaction(5)and reaction(6),respectively.This indicates that if we apply the same potential versus RHE,the driving forces for reactions(5)and(6),with H2O2as reactant,are ca.0.17 V and 0.06 V larger than that for reaction(7)with HO2−as reactant,respectively.However,the HPOOR rate we observed at lower pH is significantly smaller than that at higher pH.We think this is due to the low d-band center,which reduces the interaction between Au(111)and H2O2,hence the barrier for HPOOR through H2O2as a reactive species is very high[9].In contrast,at potentials where HPOOR occurs,the Au(111)surface is positively charged[14],and electrostatic interaction will facilitate the adsorption of HO2−.Therefore,the barrier for the oxidation of HO2−(i.e.,backward reaction of Eq.(7))will be smaller than that for oxidation of H2O2(back reaction of Eq.(5)and Eq.(6)).Under these circumstances,we think the major species to be discharged at the electrode is HO2−,which can be formed from the dissociation of H2O2near the electrode surface(reaction(1)). Hydrogen peroxide oxidation on Au(111)electrode in alkaline media(with pH from 10 to 13)has been investigated.We found that HPOOR activity increases with increasing the solution pH.HO2−is suggested to be the main reactive species for HPOOR in the alkaline media.Two possible origins may lead to the increase of HPOOR activity with solution pH increasing:(i)with the increase of solution pH,the concentrations of both reactants,i.e.,HO2−and OH−,increase.As a result,the kinetics of HPOOR increase;(ii)at the same applied potential versus RHE,the overpotential for HPOOR through reaction(7)increases with increasing pH,which will also lead to increasing HPOOR activity with increasing solution pH.The significantly smaller HPOOR current in solutions with pH=10.4 and 11.1 is probably due to the concentrations of the two reactive species of reaction(7),i.e.,HO2−and OH−are very small and the consumption of OH−shifting the interfacial pH to smaller values,which further reduces the concentration of HO2−near the electrode surface. This work was supported by the National Natural Science Foundation of China(No.21473175 and No.21273215)and the National Key Basic Research Program of China from the Ministry of Science and Technology of China(No.2015CB932301). [1]A.M.G´omez-Mar´ın,R.Rizo,and J.M.Feliu,Catal.Sci.Technol.4,1685(2014). [2]D.Mei,Z.Da He,Y.L.Zheng,D.C.Jiang,and Y.X.Chen,Phys.Chem.Chem.Phys.16,13762(2014). [3]A.Ignaczak,R.Nazmutdinov,A.Goduljan,L.M.de Campos Pinto,F.Juarez,P.Quaino,E.Santos,and W.Schmickler,Nano.Energy 29,362(2016). [4]M.F.Li,L.W.Liao,D.F.Yuan,D.Mei,and Y.X.Chen,Electrochem.Acta 110,780(2013). [5]L.W.Liao,M.F.Li,J.Kang,D.Chen,Y.X.Chen,and S.Ye,J.Electroanal.Chem.688,207(2013). [6]Y.Zheng,W.Chen,X.Q.Zuo,J.Cai,and Y.X.Chen,Electrochem.Commun.73,38(2016). [7]Y.L.Zheng,D.Mei,Y.X.Chen,and S.Ye,Electrochem.Commun.39,19(2014). [8]E.Sitta,A.M.G´omez-Mar´ın,A.Aldaz,and J.M.Feliu,Electrochem.Commun.33,39(2013). [9]Y.L.Zheng,J.Wei,and Y.X.Chen,J.Electrochem.22,602(2016). [10]A.M.G´omez-Mar´ın,A.Boronat,and J.M.Feliu,Russ.J.Electrochem.53,1029(2017). [11]J.Clavilier,R.Faure,G.Guinet,and R.Durand,J.Electroanal.Chem.Interface 211,107(1980). [12]H.Angerstein-Kozlowska,B.Conway,A.Hamelin,and L.Stoicoviciu,J.Electroanal.Chem.Interface 228,429(1987). [13]U.Zhumaev,A.V.Rudnev,J.F.Li,A.Kuzume,T.H.Vu,and T.Wandlowski,Electrochem.Acta 112,853(2013). [14]U.Hamm,D.Kramer,R.Zhai,and D.Kolb,J.Electroanal.Chem.414,85(1996).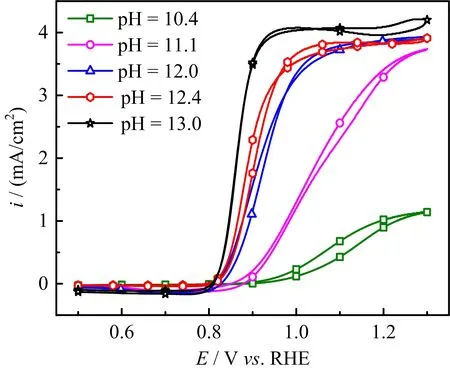
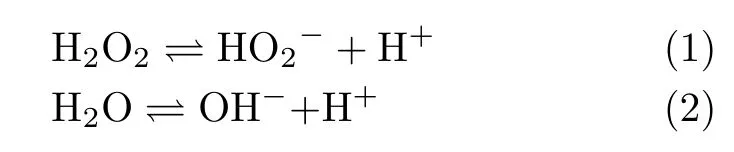
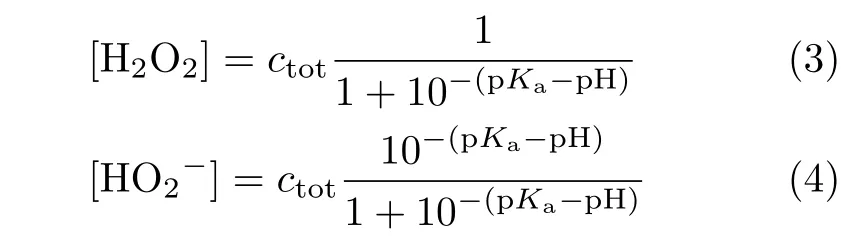
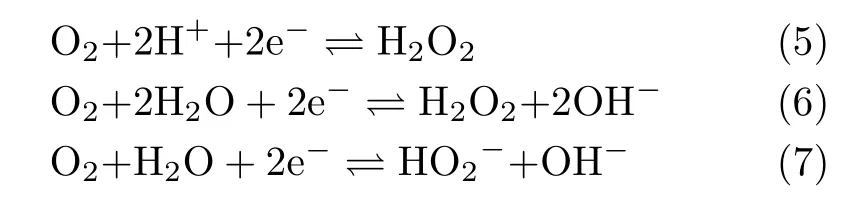
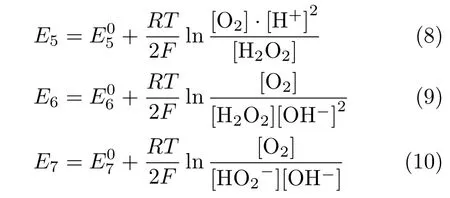
IV.CONCLUSION
V.ACKNOWLEDGEMENTS
杂志排行

CHINESE JOURNAL OF CHEMICAL PHYSICS的其它文章
- Imaging HNCO Photodissociation at 201 nm:State-to-State Correlations between CO(X1Σ+)and NH(a1∆)
- Energy-Transfer Processes of Xe(6p[1/2]0,6p[3/2]2,and 6p[5/2]2)Atoms under the Condition of Ultrahigh Pumped Power
- Ultrafast Investigation of Excited-State Dynamics in Trans-4-methoxyazobenzene Studied by Femtosecond Transient Absorption Spectroscopy
- Strong Current-Polarization and Negative Diflerential Resistance in FeN3-Embedded Armchair Graphene Nanoribbons
- Unexpected Chemistry from the Homogeneous Thermal Decomposition of Acetylene:An ab initio Study
- Direct Observation of Transition Metal Dichalcogenides in Liquid with Scanning Tunneling Microscopy