Identification of Genes Associated with Clubroot Resistance in Chinese Cabbage
2019-01-09SongBoXuHaiHeJiangmingHuJingfengFanXiaoxueandChenLongzheng
Song Bo, Xu Hai, He Jiang-ming, Hu Jing-feng, Fan Xiao-xue, and Chen Long-zheng*
1 Institute of Vegetable Crops, Jiangsu Academy of Agricultural Sciences, Nanjing 210014, China
2 Institute of Horticultura Crops, Yunnan Academy of Agricultural Sciences, Kunming 650205, China
Abstract: The transcriptome-wide gene expression was compared between susceptible and resistant Chinese cabbage cultivars to identify genes that contributed to clubroot resistance. A higher number of differentially expressed genes were detected in susceptible cultivars than in resistant cultivars. Fifty-six genes involved in cell wall modification, hormone signaling, root marphogenesis,nematodes response and cell proliferation were uniquely expressed in the resistant cultivars. Among them, 27 genes were involved in cell wall modification and hormone signaling, indicating that genes in these two types might play a vital role in the defense response to pathogen infection.
Key words: Chinese cabbage, clubroot, transcriptome analysis, resistant gene discovery
Introduction
Clubroot disease is one of the most economically important diseases of cruciferous vegetables and oil crops, such as Brassica rapa, B. oleracea and B. napus(Crute et al., 1980; Diederichsen et al., 2009; Dixon,2014) caused by the soil-borne obligate plant pathogen Plasmodiophora brassicae Wor, which can survive for up to two decades in the soil (Kuginuki et al., 1999).So far, this contagious and rapidly spreading disease is more and more resistant to traditional agricultural treatments such as liming and crop rotation (Voorrips,1995). Therefore, developing resistant cultivars is an emergency objective of cruciferous vegetable breeding programs (Diederichsen et al., 2009; Donald, 2009).Elucidating the molecular mechanism that governs the defense responses in host roots will greatly facilitate the breeding of resistant cultivars.
Most previous studies have aimed to decipher the molecular pathways underlying the plant response to P. brassicae infection. Genes involved in plant hormone metabolism, especially auxin, cytokinin and brassinosteroid in Arabidopsis thaliana will be altered,during P. brassicae infection (Schuller et al., 2014).Another microarray analysis revealed that partial resistance to clubroot is associated with reduced or delayed metabolomic changes and an earlier induction of classical defense responses (Jubault et al., 2013).Moreover, an miRNA-based analysis found that plant hormone-related genes, stress-related genes and genes encoding transcription factors are the targets of miRNAs that are differentially expressed during clubroot disease development (Verma et al., 2014).
Previous study has proved that over 2 000 genes were differentially expressed in clubroot-resistant and -susceptible Chinese cabbage cultivars. Among these genes, defense response genes, encoding enzymes associated with jasmonic acid (JA), ethylene(ET), indole glucosinolate biosynthesis and callose deposition but not those involved in the salicylic acid(SA) pathway, are up-regulated in clubroot-resistant plants, compared to susceptible plants (Chu et al.,2014). Recently, 151 genes encoding transcription factors or related to calcium ion influx, hormone signaling, pathogenesis and cell wall modifications are differentially expressed between clubroot-resistant and -susceptible cultivars after inoculation (Chen et al., 2016). Interestingly, they found that genes involved in SA signaling pathway are expressed at significantly higher levels in a clubroot-resistant cultivar than in a susceptible cultivar and predicted that SA signaling is particularly important to clubroot resistance (Chen et al., 2014). However, the data collected in these reports did not provide a comprehensive understanding of the mechanisms of resistance to clubroot in Brassica crops.
Transcriptome analysis is particularly useful for revealing the relationships between gene expression and phenotype. It has been successfully used to identify potential resistance genes for cold tolerance in spring wheat Triticum aestivum L. 'Glenlea' (Gulick et al., 2005), salt tolerance in common bean (Phaseolus vulgaris L.) (Hiz et al., 2014), drought tolerance in upland cotton (Gossypium hirsutum L.) (Bowman et al., 2013) and rice (Oryza sativa ssp. Japonica cv.Nipponbare) (Oono et al., 2014). This study compared the transcriptional responses of resistant and susceptible Chinese cabbage cultivars to P. brassicae infection. Identifying these genes and their associated pathways would improve the understanding of the mechanisms underlying clubroot resistance and provide a basis for genetic engineering strategies to develop clubroot-resistant Brassica lines.
Materials and Methods
Plant materials and inoculation
Chinese cabbage 'CRR13025', a highly resistant cultivar (RC) carrying the club root-resistant allele of CRb, was supplied by the Institute of Vegetable Crops,Yunnan Academy of Agricultural Sciences, China.Chinese cabbage 'CRS05001', a susceptible cultivar(SC), was supplied by the Institute of Vegetable Crops,Jiangsu Academy of Agricultural Sciences, China.After sterilization, seeds were germinated at 30℃.Uniformly germinated seeds were collected and sown in sterilized soil and cultured in the greenhouse at 25℃/15℃, with a photoperiod of 16 h light/8 h darkness. After four true leaves had emerged, the seedl-ings of the resistant and susceptible cultivars were divided into two groups. One group was inoculated by dipping the roots into a 1×106spores • mL-1suspension of an isolate of P. brassicae and the other served as the control and was inoculated with distilled water in the same way. Roots of 10 individual plants from each group were harvested 8 days after inoculation and rapidly stored at –80℃ until using. Three biological replicates were used for each group. The remaining plants were assessed 28 and 50 days after inoculation to establish whether or not the inoculation was successful.
RNA isolation and library construction for Illumina sequencing
Total RNA was extracted from root tissues of the resistant and susceptible cultivars using Trizol Reagent(Invitrogen, CA, the USA), according to the manufacturer's instructions. The quality and integrity of RNA were determined using a NanoDrop spectrophotometer (NanoDrop, the USA) and an Agilent 2100 Bioanalyzer (Agilent, USA), respectively. The mRNA was converted to a library for subsequent cluster generation using an Illumina TruSeq RNA Sample Preparation Kit (Illumina, the USA). mRNA was purified using magnetic beads attached to poly-Toligo. Subsequently, mRNA was fragmented into small pieces using divalent cations under elevated temperature. Thefirst-strand cDNAs were generated from the cleaved RNA fragments using reverse transcriptase and random primers. The second-strand cDNAs were synthesized using DNA polymerase I(Fermentas, USA), and RNase H was used to degrade the template RNA. A paired-end library was constructed from the synthesized cDNA using the Genomic Sample Prep Kit (Illumina). The cDNA fragments were purified using QIAquick PCR Extraction Kit (Qiagen, the USA) and were then ligated with adapters as reported previously (Margulies et al.,2005). Excess adapters and adapter dimers were removed using AMPureXP beads and a sequencing library was constructed using polymerase chain reaction (PCR) amplification. The multiplexed DNA libraries were normalized to 10 nmol • L-1and pooled in equal volumes. Two normalized cDNA libraries were constructed using RNA from inoculated plants and the mock-inoculated control. The libraries were sequenced using Illumina Hiseq 2000 platform(Shanghai Personal Biotechnology Co., Ltd., China).
Datafiltering
Paired-end, 100 bp raw reads were generated using RNA-Seq. The reads were screened from the 3' to 5'end to trim the bases with a quality score (Q) of < 20 using 5 bp windows. Reads with afinal length of<50 bp were removed. After the adaptor sequences were removed, low-quality reads with ambiguous'N' nucleotides (with an 'N' ratio of more than 10%)and low quality sequences (with Q<5) were removed.
Gene annotation, gene expression and functional enrichment analyses
The reference sequences were indexed by Bowtie with a Burrows-Wheeler index which could keep its memory footprint small (Langmead et al., 2012). The sequencing reads were mapped to B. rapa genome(ftp://brassicadb.org/Bra_Chromosome_V1.5/) using TopHat (Trapnell et al., 2009). EBSeq was used to identify DEGs. DEGs werefiltered using a false discovery rate below 0.01. The genome annotation was download form the Brassica Database. The genes from the annotation were compared with the non-redundant protein (nr) database (Deng et al., 2006). The Cluster of Orthologous Groups (COG) database (Galperin et al., 2000), the Kyoto Encyclopedia of Genes and Genomes (KEGG) Pathway database and Gene Ontology (GO) (Kanehisa et al., 2004; Ball et al.,2000). The Blast2GO program was used to obtain GO annotation of the genes (Conesa et al., 2005). TopGO software was then used to perform GO enrichment analysis of differentially expressed genes. WEGO(http://wego.genomics.org.cn/cgi-bin/wego/index.pl)was used to produce GO functional classification of all the expressed genes. At last, the pathway enrichment analysis of DEGs was performed by hypergeometric test.
Validation of differentially expressed genes by quantitative PCR
The expression of eight randomly selected differentially expressed genes was validated and quantified using quantitative PCR (qPCR). Primers (Table 1)were designed, according to Illumina sequencing data using Primer 5.0 (PREMIER Biosoft International, CA, the USA). Reverse cDNA was synthesized using the PrimeScriptTM RT Reagent Kit with gDNA Eraser (TaKaRa). qPCR was performed using an Eppendorf Mastercycler ep Realplex. The actin gene of Chinese cabbage was used as an internal control to normalize the expression level and all the experiments were performed in triplicate. The reaction was carried out in a total volume of 20 μL containing 10 μL 2×SYBR real-time PCR Pre-mix (TaKaRa),1 μL diluted cDNAs, and 0.4 μL of each primer(10 mmol • L-1). The thermal cycler profile for SYBR Green qPCR was 95℃ for 4 min, followed by 35 cycles of 95℃ for 15 s, 57℃ for 15 s, and 72℃ for 25 s.Amplification and detection of only one PCR product was confirmed by performing melting curve analysis of the amplification products at the end of each PCR cycle. After PCR program, the expression level of different genes was analyzed using the 2-ΔΔCTmethod (Livak et al., 2001).
Results
RNA sequencing of Chinese cabbage transcriptome
Clubs could only be detected in inoculated samples of the susceptible cultivars (SC-T) 28 and 50 days postinoculation (dpi), while no clubs were found in the control samples (SC-CK), inoculated resistant cultivars(RC-T) and the control samples (RC-CK) ( Figs. 1, 2).Two treated samples (SC-T and RC-T) and the two controls (SC-CK and RC-CK) were sequenced and compared to a reference genome of Chinese cabbage(ftp://brassicadb.org/Bra_Chromosome_V1.5/). The expression levels of the genes were similar among the four samples, suggesting that there was no bias in construction of the cDNA libraries (Fig. 3).
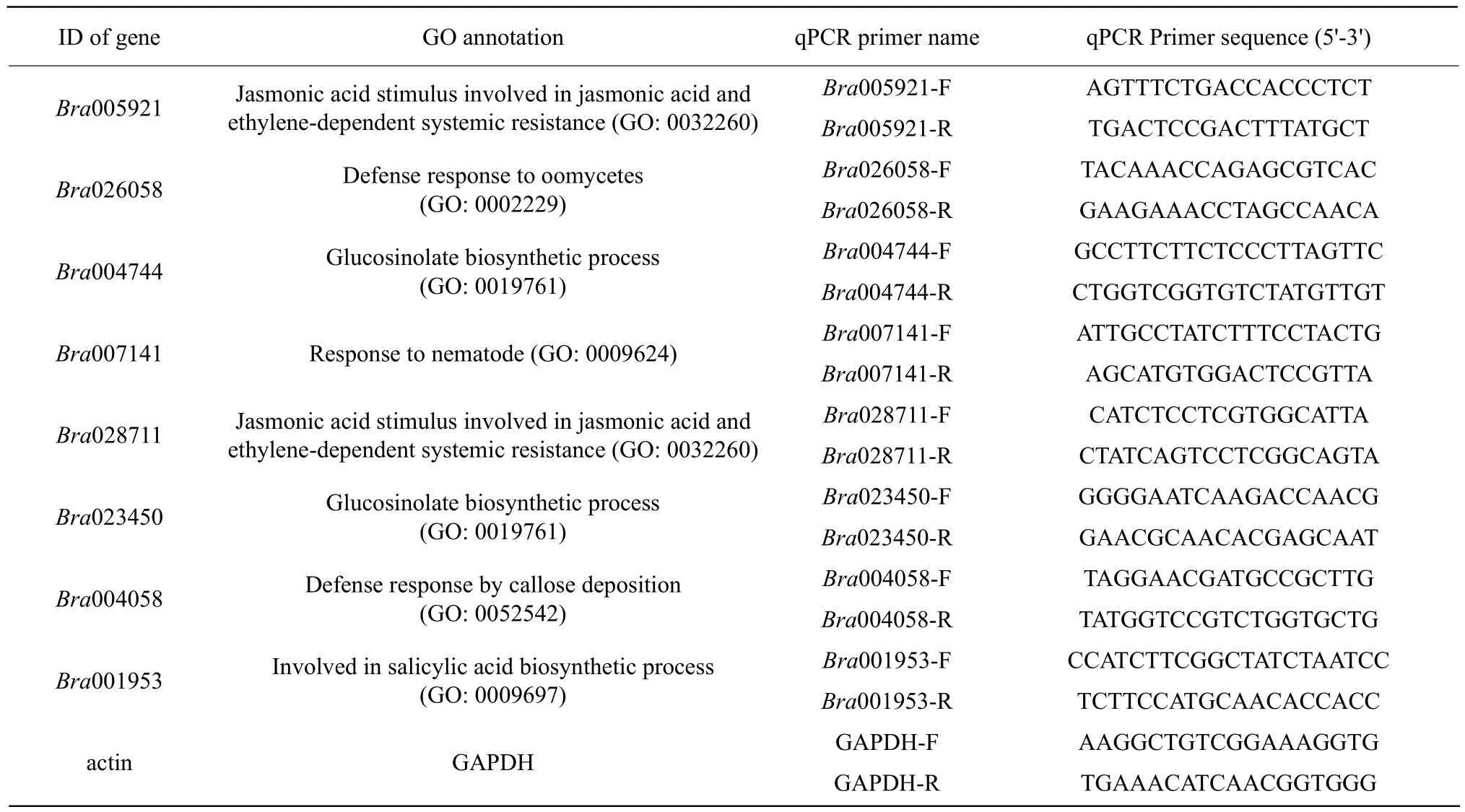
Table 1 Genes and specific primers used for real-time polymerase chain reaction

Fig. 1 Visible clubs on SC-T inoculated after 28 days (shown by arrow)

Fig. 2 More visible clubs on SC-T inoculated after 50 days (shown by arrow)
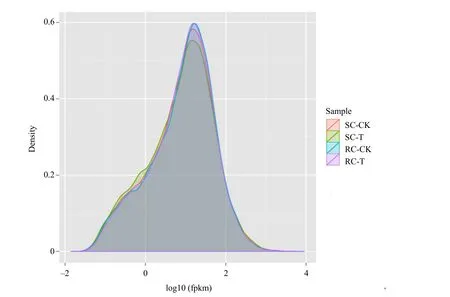
Fig. 3 Expression level distribution of all the unigenes among the four libraries
The total mapped reads were aligned to each region in the reference genome, including the exon, intergenic and intron regions, with the percentage of the exon region being the highest in all the four samples libraries (Fig. 4). GC% of all the four sequences libraries was approximately 47% and Q30 percentage was above 88.52% (Table 2). A total of 13 405 116 and 13 018 837 clean reads were obtained from SC-T and SC-CK, respectively; 12 637 292 and 13 811 534 clean reads were obtained from RC-T and RC-CK,respectively. The mapped ratios of these four samples to the reference database for B. rapa genome were all above 80% (Table 2). These results suggested that the sequence data were reliability.
Detection of differentially expressed genes
Numerous differentially expressed genes (DEGs)were detected in the comparisons of plants subjected to the control and inoculation treatments. In total,1 494 DEGs were detected between the control and the inoculated treatments in SC, while only 753 DEGs were detected in RC, indicating that fewer changes in gene expression occurred in RC in response to inoculation than in SC. The result of DEGs hierarchical clustering is shown in Fig. 5. The similar expression pattern between SC-CK and SC-T and the similar expression pattern between RC-CK and RC-T were exhibited by hierarchical clustering analysis.
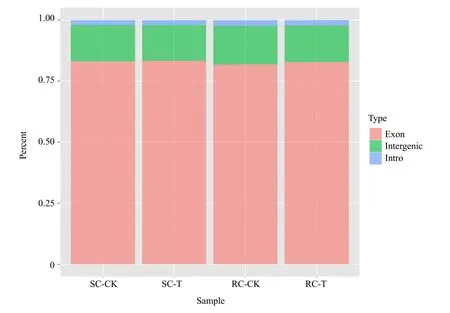
Fig. 4 Percentage of Illumina sequencing reads mapped to reference genome regions among the four libraries
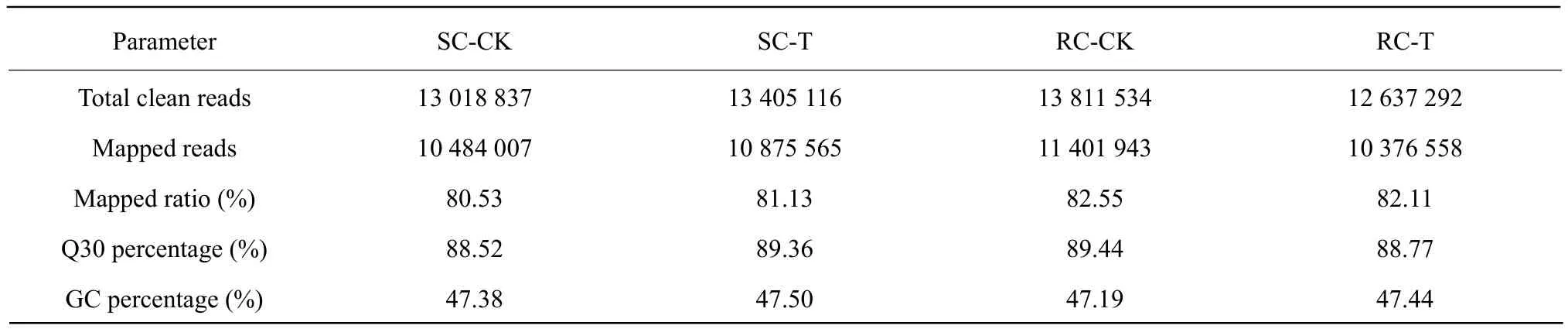
Table 2 Summary of Chinese cabbage transcriptome
Among 1 494 DEGs in SC upon inoculation, 1 310 were detected only in SC and 184 were shared with RC. Furthermore, 734 of the 1 310 DEGs (56.0%)in SC were up-regulated, which was greater than the numbers of down-regulated DEGs (576; 44.0%)(Table 3). In RC, 559 DEGs were unique to RC and 184 were shared with SC. However, in contrast to SC, only 216 of 559 DEGs (38.6%) in RC were upregulated and 343 DEGs (61.4%) were down-regulated(Table 3), which suggested that fewer genes were upregulated in RC than in SC. These results suggested that RC might not have to up-regulate genes to resist pathogen infection.
As stated previously, 184 DEGs were shared between SC and RC; 40 of them were identically up-regulated and 48 were identically down-regulated (Table 3).The data also showed that 64 of these shared DEGs were up-regulated in SC, but down-regulated in RC;32 genes were up-regulated in RC, but down-regulated in SC (Table 3), which meant that the numbers of up-regulated genes were lower in RC than those in SC.

Fig. 5 Hierarchical clustering analysis of DEGs based on RPKM data
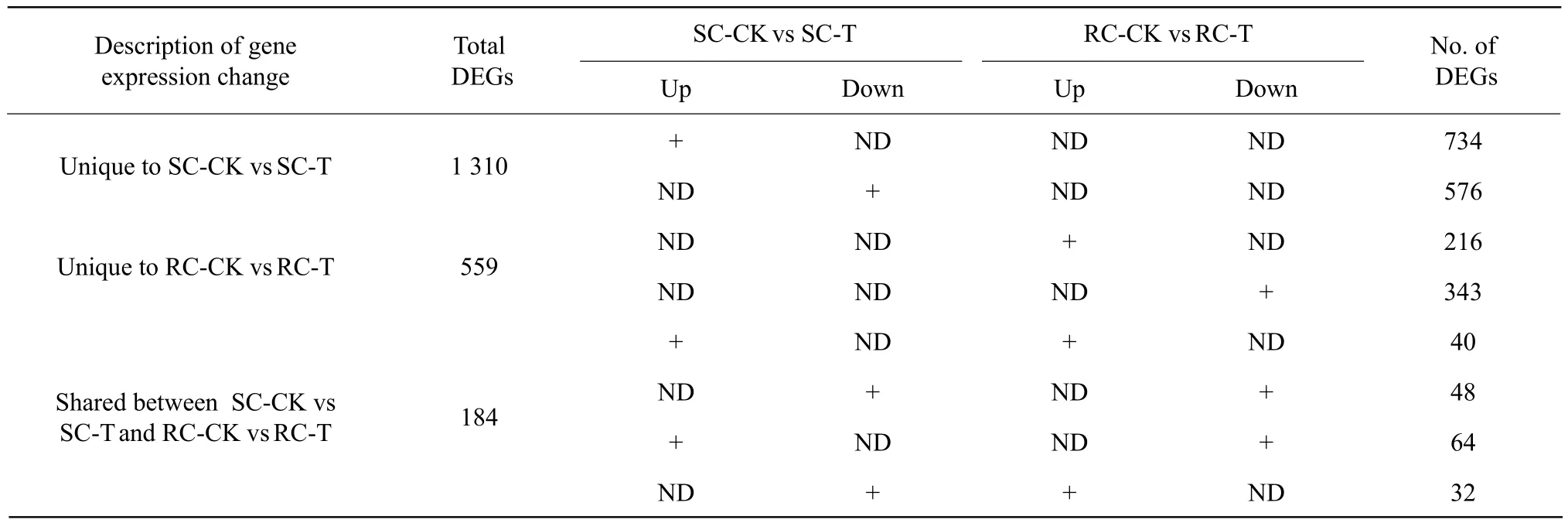
Table 3 Number of differentially expressed genes detected in sequence analysis
The gene that was up-regulated to the greatest extent (ID: Bra005921, log2FC=7.34) in RC encoded a protein with flavonoid sulfotransferase activity (GO:1990135), which was important for plant growth,development and disease prevention. Another gene(ID: Bra023450, log2FC=6.23), which encoded a protein involved in the biosynthesis of glucosinolates,compounded with antifungal and antibacterial properties (GO: 0019761), was up-regulated to a greater extent in RC than in the SC (log2FC=1.08). The gene that was down-regulated to the greatest extent in RC (ID: Bra027201, log2FC=–5.33) was a negative regulator of defense responses (GO: 0031348) and was significantly up-regulated in SC (log2FC=3.75). These DEGs might play an important role in clubroot resistance in both RC and SC.
DEGs uniquely up-regulated in resistant cultivars
To gain insight into the defense mechanisms of RC, 559 DEGs that were unique to RC, including 216 up-regulated DEGs and 343 down-regulated DEGs, were further analyzed. Among 216 upregulated DEGs, 56 were greatly raised (log2FC≥2)(Table 4) and might play a vital role in clubroot resistance.
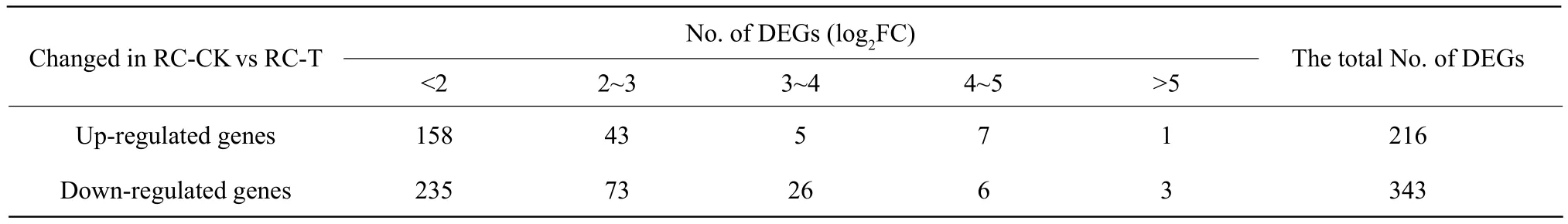
Table 4 Number of DEGs uniquely detected in resistant cultivars
Categorical classification of these 56 DEGs with significantly up-regulated expression (log2FC≥2) in RC during pathogen infection related to: (1) cell wall modifications (14 DEGs, 25.0% of the total), including 11 DEGs (Bra000002, Bra040486, Bra008796,Bra008870, Bra022002, Bra012676, Bra020322,Bra026260, Bra029172, Bra030619 and Bra030702)involved in cell wall structural constituents, organization, or biogenesis; two DEGs (Bra026058 and Bra032734) related to the defense response by callus deposition in the cell wall; and one DEG (Bra011292)related to the lignin biosynthetic process; (2) the response, biosynthesis and metabolism of plant hormones(18 DEGs, 32.1% of the total), includingfive DEGs(Bra003817, Bra038726, Bra009300, Bra028711 and Bra000760) involved in jasmonic acid- and ethylene-dependent systemic resistance; three DEGs(Bra000265, Bra003588 and Bra034572) related to the salicylic acid-mediated signaling pathway and systemic acquired resistance; four DEGs (Bra004744,Bra013011, Bra017914 and Bra029355) related to glucosinolate biosynthesis; two DEGs (Bra011490 and Bra026465) related to brassinosteroid metabolism;and four DEGs (Bra037177, Bra029481, Bra015999 and Bra039265) related to the abscisic acid-activated signaling pathway; (3) two DEGs (Bra000129 and Bra008796) related to root morphogenesis; (4) two DEGs (Bra007141 and Bra021374) related to the plant's response to nematodes; and (5) 20 DEGs related to the nucleus, membranes, intracellular signal transduction and others (Table 5). It was proposed that these uniquely up-regulated DEGs and their associated metabolic pathways might play important roles in the defense response to pathogen infection.
Validation of expression patterns by qPCR analysis
To validate RNA-seq results, qPCR was performed on the following eight randomly selected DEGs that were significantly up-regulated in response to infection: Bra005921 (encoding an enzyme with flavonoid sulfotransferase activity; GO: 1990135);Bra028711 (jasmonic acid stimulus, jasmonic acid and ethylene-dependent systemic resistance; GO: 0032260,Table 5); Bra023450 and Bra004744 (glucosinolate biosynthesis; GO: 0019761); Bra007141 (nematode resistance; GO: 0009624, Table 5); Bra026058(defense response by callose deposition in the cell wall; GO: 0052544, Table 5); Bra004058 (defense response by callose deposition; GO: 0052542); and Bra001953 (systemic acquired resistance, salicylic acid-mediated signaling pathway; GO: 0009862). The results indicated that the expression of these eight DEGs was up-regulated dramatically, during infection(Fig. 6), which corroborated thefindings of RNA-seq analyses.
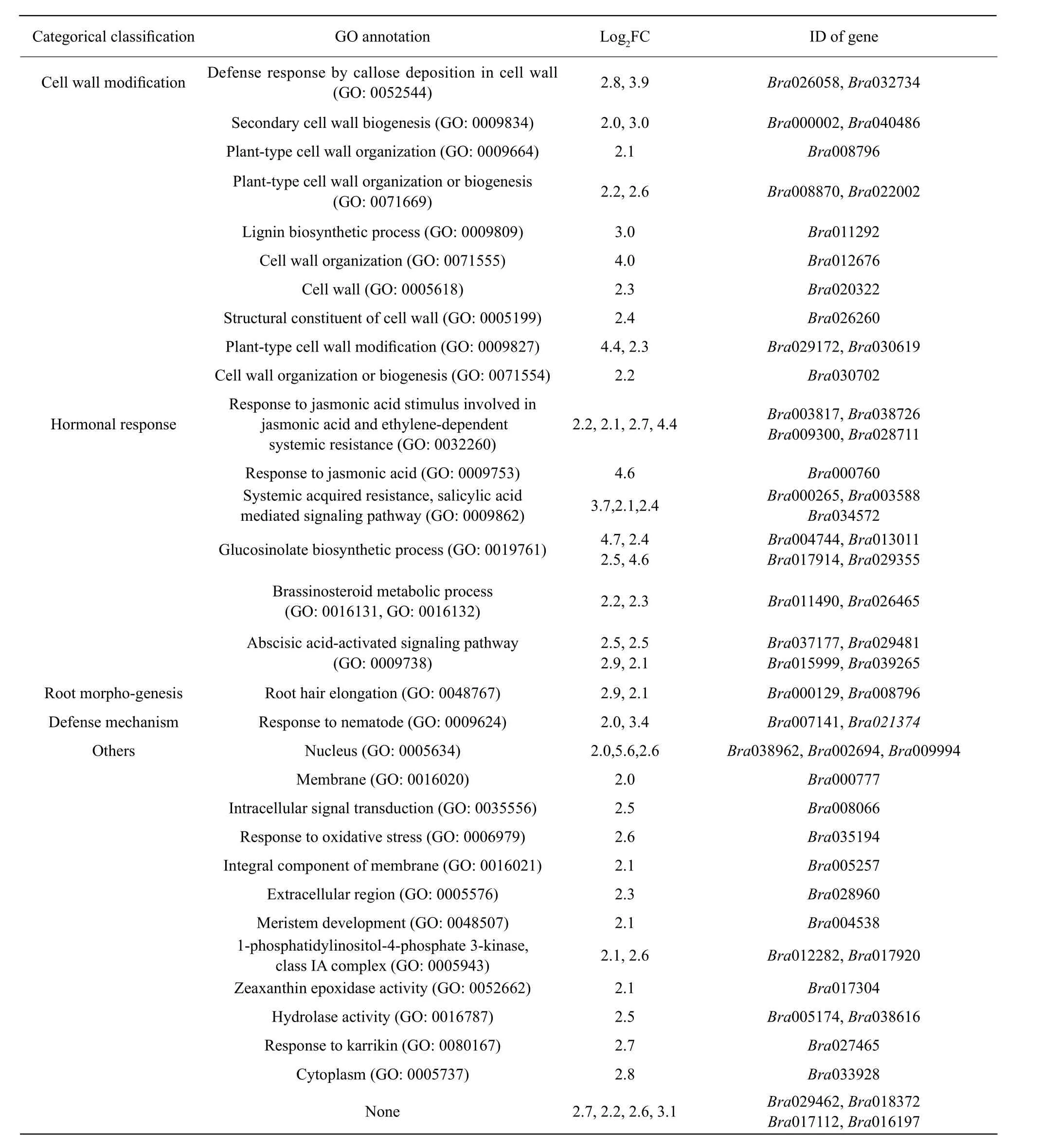
Table 5 DEGs uniquely up-regulated in resistant cultivars with annotated functions
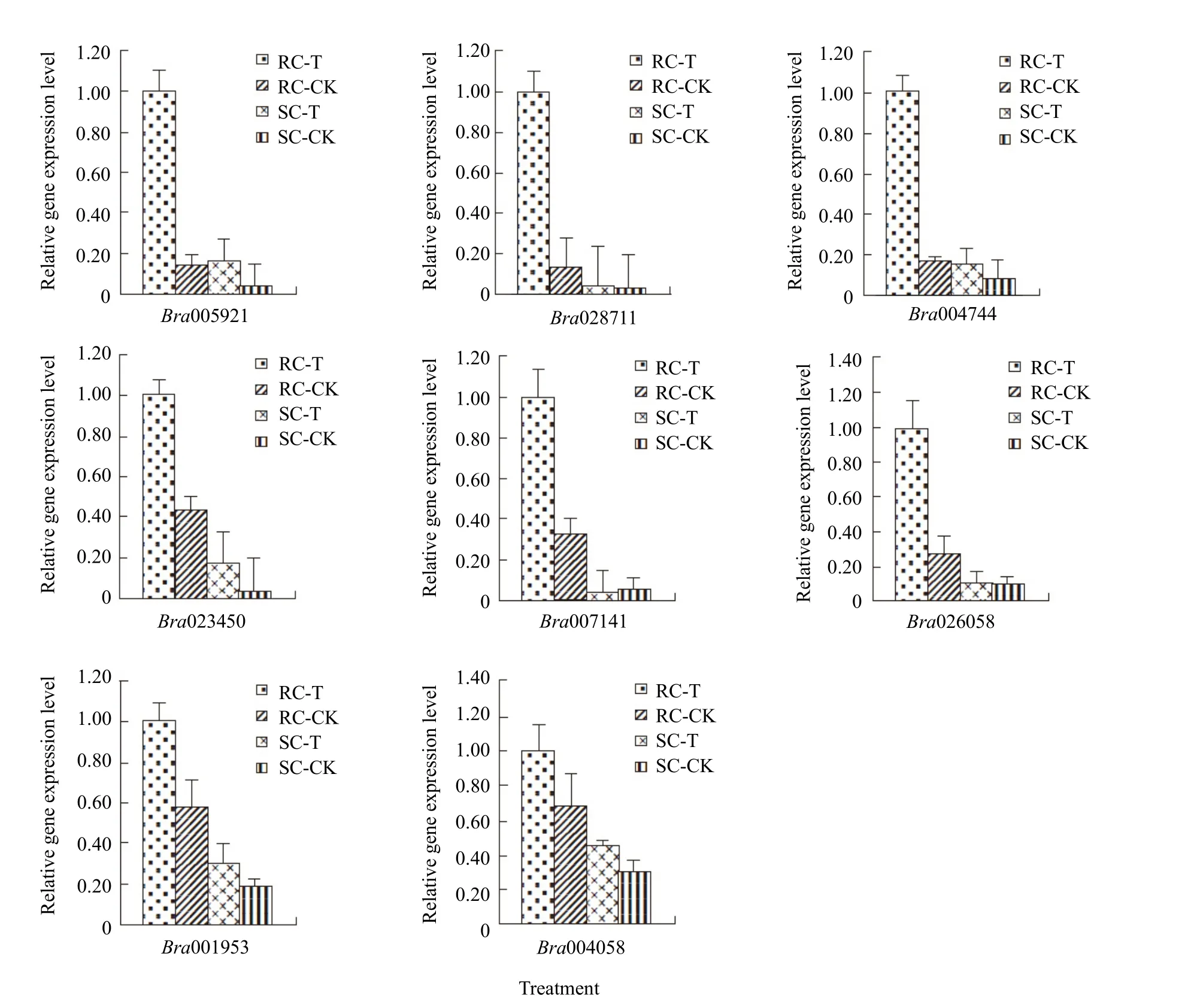
Fig. 6 Relative expression level of eight DEGs among pathogen inoculated and control Chinese cabbage obtained by qPCR
Discussion
The stability of a stress responsive gene expression network might play an important role in the defense response to environmental stresses. In this study, it was found that there were fewer instance of altered gene expression upon P. brassicae infection in RC than in SC. Moreover, the numbers of up-regulated DEGs were much smaller in RC than that in SC.Therefore, it was hypothesized that it might not be as necessary for RC to modulate gene expression networks to defend against pathogen infection as it was in susceptible cultivars.
Jasmonic acid (JA) played an important role in the defense against bacterial and fungal infections,insect attack and the wound response. (Chu et al.,2014; Glazebrook, 2005; Pieterse et al., 2012; Shin et al., 2016). Previous study had proved that the signaling and metabolic activity of JA were upregulated significantly in resistant populations of Chinese cabbage compared to the susceptible lines at 15 days post-inoculation with P. brassicae (Chu et al.,2014); specifically, the signaling network primarily regulated by exogenous JA has been shown to trigger a cascade of events that led to enhanced plant resistance to pathogens (Glazebrook, 2005; Pieterse et al., 2012;Robert-Seilaniantz et al., 2011; Pieterse et al., 2009).Recent study showed that JA/ET signaling pathway did not play a critical role in host resistance to P. brassicae infection in Chinese cabbage as all the 17 DEGs related to JA/ET signaling pathway were down-regulated in the resistant cultivars (Chen et al.,2016). In this study,five DEGs involved in JA metabolism were significantly up-regulated in the resistant cultivars, indicating that the signaling and metabolic activity of JA might play an important role in the defense response to clubroot.
Many studies have demonstrated that plant resistance to biotrophic pathogens was controlled largely by SA-mediated signaling pathways (Glazebrook,2005). In Arabidopsis, SA treatment had a protective effect against clubroot symptoms (Agarwal et al.,2013; Jubault et al., 2008) which could be triggered by P. brassicae infection in the Bur-0 accession (Fahey et al., 2001). Recent study showed that SA signaling pathway played a critical role in host resistance to P. brassicae infection of Chinese cabbage (Chen et al.,2016). Taken together, these studies suggested that SA accumulation in the host could facilitate resistance to P. brassicae. However, the involvement of salicylic acid (SA) in the defense response to pathogen infection had not been fully clarified. Although in previous study, no significantly different expressed genes were detected involved in SA metabolic and signaling pathways, no matter in clubroot-resistant and -susceptible Chinese cabbage (Chu et al., 2014)or in Glycine max (soybean) (Hamon et al., 2008). In this study, it was found that three of the 56 DEGs in RC that were significantly up-regulated in response to pathogen infection, compared to SC, were involved in SA-mediated signaling pathways, indicating that SA-mediated signaling pathways were important for resistance to infection of P. brassicae. Together, the results indicated that both JA signaling pathway and SA signaling pathway played a critical role in host resistance to P. brassicae infection.
Glucosinolates were the major class of secondary metabolites found in Brassicaceae species (Kim et al.,2011; Fahey et al., 2001). In plants, the glucosinolate system acted as an efficient defense mechanism against environmental stresses, such as predation by herbivores, microorganisms, fungi, wounding and chemical toxicity (Agerbirk et al., 2009; Hopkins et al., 2009); Bednarek et al. (2009) reported that glucosinolates were recruited for broad-spectrum antifungal defense responses in plants. Glucosinolate levels in oilseed rape were positively correlated with resistance to pathogens (Li et al., 1999) and several glucosinolate hydrolysis products had been reported to display toxicity to fungi and bacteria (Mayton et al.,1996; Brader et al., 2001). In this study, four significantly up-regulated DEGs involved in the glucosinolate biosynthesis were uniquely detected in the resistant cultivars, including Bra004744, Bra013011,Bra017914 and Bra029355. These genes expression products might play a role in limiting the infection by P. brassicae in the resistant cultivars.
The plant cell wall developed a physical barrier to the entry of various biotic attackers. When attacked,a modification of the cell wall secreted several compounds that could trigger defense responses and provide protection to the plant (Cantu et al., 2008).Previous studies demonstrated that cell wall modifications, such as lignification, cell division and expansion, played important roles during the defense response to V. dahliae in Gossypium hirsutum (cotton)(Toyoda et al., 2016; Smit et al., 1997; Mcfadden et al., 2001). Siemens et al. (2006) reported that genes involved in cell division and expansion, such as cell cycle genes and expansins, were up-regulated in A. thaliana after inoculation with P. brassicae. Of the 56 uniquely up-regulated genes in RC in the study, 14 were involved in cell wall modification (specifically,plant-type cell wall organization or biogenesis, callus deposition in the cell wall, positive regulation of cell proliferation, and lignin biosynthesis), indicating that these genes might contribute to resistance.
The defense responses of plants were regulated by multiple signaling pathways and there was significant overlap between the patterns of gene expression that were induced in plants in response to different stresses(Durrant et al., 2000; Seki et al., 2001; Singh et al.,2002; Kliebenstein, 2016). For example, the gene expression profiles observed during an incompatible plant-fungal interaction overlapped with those derived from wounding (Durrant et al., 2000). Using cDNA microarrays, a substantial number of genes had been found to be coordinately regulated by different defense/stress signals, by infection with a fungal pathogen (Schenk et al., 2000) or by cold or drought stress (Seki et al., 2001). For instance, two genes that were up-regulated in response to P. brassicae in the study, Bra007141 and Bra021374, were also involved in the response to nematodes, suggesting these genes might be coordinately regulated by nematode and fungal infection in roots.
Conclusions
This transcriptome analysis focused on Chinese cabbage cultivars that were resistant or susceptible to clubroot and the results suggested that a stable gene expression network response to pathogen infection might play an important role in the defense responses.The defense response of plants was not determined solely by one gene or one factor, but depended on a variety of factors, such as the timing of recognition,the activity of pathogen effectors, and the type of plant tissue. These different factors together shaped the nature of the specific defense response of the plant. Therefore, more studies are needed to clarify the resistance mechanisms of Chinese cabbage. In this study, 56 genes that were differentially expressed in a RC of Chinese cabbage upon P. brassicae infection were identified and classified. Thesefindings contribute to our understanding of the molecular mechanisms underlying the resistance of B. rapa to P. brassicae infection.
杂志排行

Journal of Northeast Agricultural University(English Edition)的其它文章
- Laboratory Assessment of Susceptibility of Maize Varieties to Postharvest Infestation By Sitophilus zeamais (Mostchulsky) (Coleoptera:Curculionidae)
- Species Composition and Diversity Analysis of Lava Flow in Different Periods in Wudalianchi Nature Reserve, China
- FSH Promoting Proliferation of Calf Sertoli Cells Through Wnt/β-catenin Signaling Pathway with CDC25B Being Involved
- Function of RanGAP1 in Mouse Oocyte Fertilization
- Porcine Parvovirus Inducing Autophagy to Benefit Its Replication
- Isolation and Characterization of E. Coli O157 : H7 from Infected Newborn Calves in Northeast China