钠离子对硅酸二钙碳化产物的影响
2018-12-27刘松辉张海波管学茂张富东魏红姗
刘松辉, 张海波, 管学茂, 张富东, 魏红姗
(河南理工大学 材料科学与工程学院, 河南 焦作454003)
二氧化碳的大量排放带来了一系列的环境问题,探索有效的二氧化碳减排技术是国内外学者研究的热点.在众多二氧化碳减排技术中,二氧化碳的矿物封存被认为是最有前景的封存途径[1-3].此外,水泥行业排放的二氧化碳占全球人类活动排放二氧化碳总量的5%,因此开发新一代绿色低碳胶凝材料也是国内外研究的热点[4-6].
众所周知,大部分硅酸钙矿物(包括C3S,C2S,C3S2和CS等)在热力学上是能够与二氧化碳反应的.利用硅酸钙矿物与二氧化碳的碳化反应不仅能够实现二氧化碳的矿物封存,同时能够形成致密的硬化体结构.笔者在文献[5]中介绍了通过激发γ-C2S矿物碳化活性来制备绿色低碳胶凝材料的方法,发现γ-C2S矿物加速碳化8h后的抗压强度可达71.3MPa,其碳化生成的方解石晶体和高度聚合的SiO2凝胶(类似于C-S-H凝胶)是强度增加的主要来源.
碳化产物的结构与胶凝材料的性能密切相关.然而硅酸钙碳化产物结构复杂,国内外学者对硅酸钙碳化产物的结构(包括晶态结构和非晶态结构)尚没有统一的结论[4-11],影响机理尚不清晰,甚至多数研究没有涉及非晶态碳化产物结构的表征[6,8-11].此外,一些工业废弃物(如钢渣、赤泥等)含有大量的硅酸钙矿物,通过激发这些工业废弃物的碳化活性来制备建筑制品是一种应用前景巨大的技术[10-11],但是这些工业废弃物除了含有硅酸钙矿物外,还含有钠、钾等碱金属离子,而关于这些杂质离子对硅酸钙矿物碳化反应的影响以及可溶碱固化方面的研究还很缺乏.
本文制备了β-C2S单矿,通过内掺不同质量分数的NaCl来引入钠离子,研究了钠离子对β-C2S矿物碳化产物结构、碳化程度和微观形貌的影响规律,探索了利用β-C2S矿物碳化来固化可溶性钠离子的可行性,为开发新一代绿色低碳水泥胶凝材料以及工业废弃物的绿色利用提供理论依据.
1 试验
1.1 β-C2S的制备
原材料碳酸钙、二氧化硅、氧化钡、NaCl等均为分析纯,购自上海麦克林生化科技有限公司.
1)文中涉及的含量、比值等均为质量分数或质量比.
按照硅酸二钙(C2S)的化学组成称取碳酸钙和二氧化硅进行混合,外掺质量分数为0.3%的氧化钡,混匀、磨细、压制成块.在高温炉内于1350℃下煅烧 2h.然后快速冷却(冷却速度>500℃/min),再重新压制成块进行煅烧,如此重复,直到制得的样品用乙醇-乙二醇法测得其f-CaO含量1)小于1.5%.所得样品经粉磨后,采用马尔文激光粒度仪测得平均粒径d50=42.2μm,勃氏法比表面积为4100cm2/g.采用Rietveld全谱拟合法对所得样品进行定量分析,结果表明其β-C2S质量分数为92.4%.
1.2 加速碳化
将制备好的β-C2S矿物分别内掺质量分数为0%,5%,10%的中性钠盐NaCl(其他钠盐如NaOH,Na2CO3,NaHCO3等在引入钠离子的同时会改变液相pH值,影响碳化反应),样品记为N-0,N-5和N-10.控制水料比为0.1以利于碳化反应.将以上混合料分别与水混合均匀后放入碳化反应釜,先将反应釜抽真空到-0.1MPa,维持10min后,将CO2气体从气瓶通过减压阀通入反应釜内,气瓶内CO2气体浓度为99%;控制反应釜内CO2压力为0.3MPa,在室温条件下,分别碳化1,24h,然后取出样品,烘干、粉磨,测试样品的性能.
1.3 测试方法
采用德国布鲁克AXS有限公司的D8ADVANCE型X射线衍射仪(XRD)分析样品的晶体结构,Cu靶,扫描速率10(°)/min,扫描范围10°~70°,采用Rietveld全谱拟合法对样品进行定量分析.采用BJ-HCT-3综合热分析仪(TG-DTA)测试样品的TG-DTA曲线,升温速率10℃/min,测试范围50~1000℃.采用V70全自动傅里叶变换红外光谱仪(FTIR)测试样品的红外光谱,KBr压片法,测试范围400~2000cm-1.用FEI Quanta 250型场发射环境扫描电镜(SEM)和布鲁克Quantax 200型X射线能谱仪(EDS)观察样品形貌和结构,样品测试前喷金处理.由于C2S早期水化速率非常缓慢,由其水化产生的可溶性离子浓度与所掺NaCl溶解产生的离子浓度相比可以忽略不计,因此各样品中可溶性钠离子的浓度可用样品悬浮液的电导率来间接表征.用上海三信MP522型精密pH/电导率仪测试相同质量浓度(均为0.5g/L)下各样品悬浮液的电导率,溶剂为蒸馏水,测试时间统一为1min.
2 结果与讨论
2.1 XRD分析
各样品的XRD图谱及Rietveld法定量分析结果如图1和表1所示.从图1(a)可以看出:实验室制备的β-C2S矿物(用N-0-0h表示)与β-C2S矿物标准卡片(PDF#33-0302)相匹配;样品N-0经24h碳化后,方解石(calcite,PDF#05-0586)的衍射峰强度明显增强,β-C2S矿物的衍射峰强度则大大降低.结合定量分析结果(表1)可见,样品N-0经24h碳化后生成了大量的方解石晶体和少量的球霰石晶体(vaterite,PDF#33-0268),且方解石占总碳酸钙(方解石+球霰石)含量的77.4%,碳化程度为58.5%.从图1(b),(c)可以看出,在内掺NaCl的条件下,β-C2S矿物碳化产物的晶体结构发生了明显变化,球霰石晶体的衍射峰明显增强.结合定量分析结果(表1)可知:在内掺5%和10%NaCl的条件下,样品N-5和N-10碳化24h后生成的方解石只占总碳酸钙含量的11.1%和20.5%,碳酸钙主要以球霰石晶体的形式存在.由表1还可见,在相同碳化时间下,随着NaCl掺量的增加,β-C2S矿物的碳化程度没有明显差异.由图1还可发现:在相同NaCl掺量下,随着碳化时间的延长,NaCl晶体的衍射峰强度明显减弱,说明碳化后NaCl可能以非NaCl晶体的形式被固化,并且随着碳化时间的延长,NaCl的固化量提高.综上可知,NaCl影响β-C2S矿物碳化后碳酸钙晶体的结构,但不影响其碳化程度,且NaCl的固化量与碳化产物含量密切相关.

图1 各样品的XRD图谱Fig.1 XRD patterns of samples
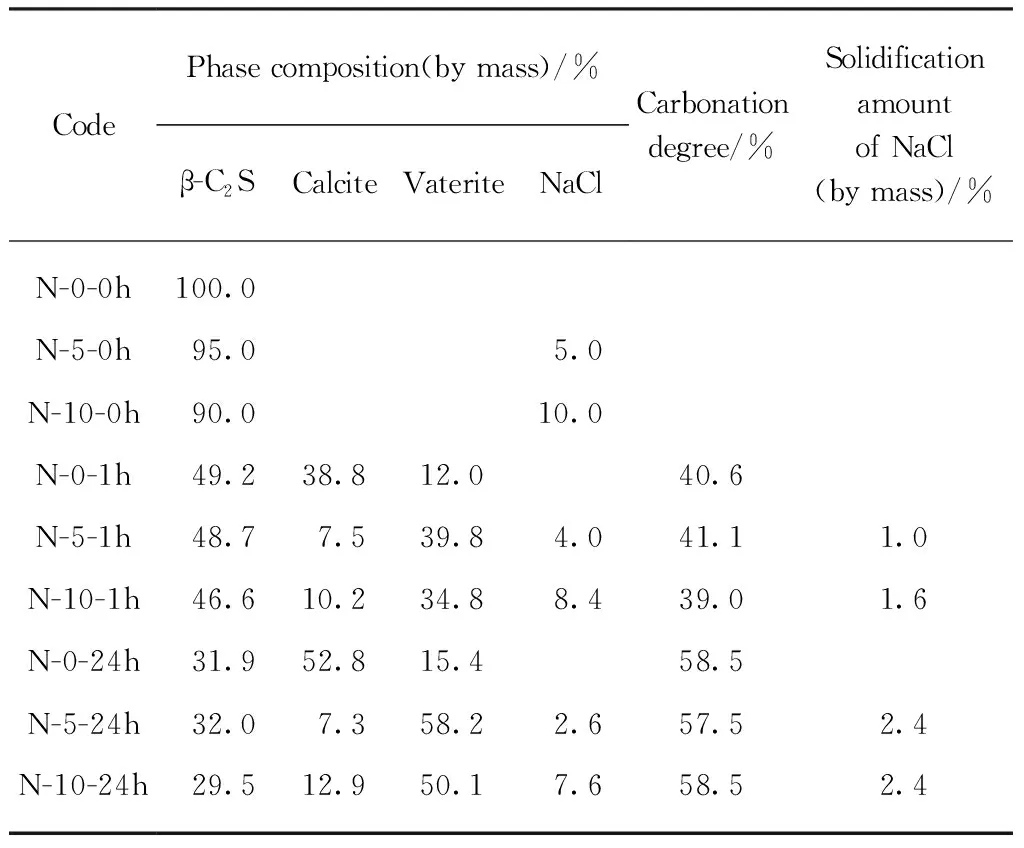
CodePhase composition(by mass)/%β-C2SCalciteVateriteNaClCarbonation degree/%Solidification amount of NaCl(by mass)/%N-0-0h100.0N-5-0h95.05.0N-10-0h90.010.0N-0-1h49.238.812.040.6N-5-1h48.77.539.84.041.11.0N-10-1h46.610.234.88.439.01.6N-0-24h31.952.815.458.5N-5-24h32.07.358.22.657.52.4N-10-24h29.512.950.17.658.52.4
2.2 TG-DTA分析
为了进一步验证以上结果,测试了各样品的TG-DTA曲线,如图2所示.从图2(a),(b)可以看出:β-C2S矿物碳化后在791℃附近出现强吸热峰,并伴随失重,表明方解石晶体发生分解并释放二氧化碳;随着碳化时间的延长(从1h到24h),失重率从10.65%增加到15.90%.由图2(c),(d)可见:在相同碳化时间下,相比样品N-0,样品N-5的TG曲线在500~850℃区间内的失重率(总碳酸钙含量)没有明显变化,说明NaCl不影响β-C2S矿物的碳化程度;但是,样品N-5的DTA曲线发生了明显变化,说明在内掺NaCl的条件下,碳化产物的组成结构发生了明显变化.在594℃附近吸热并伴随失重的过程为球霰石晶体吸热释放二氧化碳的过程;在670℃附近的吸热过程为β-C2S矿物吸热转化为α′-C2S的过程;在721℃附近的吸热过程为NaCl晶体的吸热熔化过程,且该熔化过程伴随着质量缓慢下降[12],这也是样品N-5的TG曲线在850℃以后还在缓慢下降的原因.虽然由于NaCl熔化造成质量缓慢下降,使得通过计算500~850℃区间内的失重率来定量表征碳化程度的方法有一定偏差,但是以上试验结果很好地验证了XRD的测试结果,即NaCl只影响β-C2S矿物碳化产物的组成结构,而不影响其碳化程度.

图2 各样品的TG-DTA曲线Fig.2 TG-DTA curves of samples
2.3 FTIR分析
前文采用XRD和TG-DTA对β-C2S矿物碳化产物中晶体碳酸钙的含量和结构进行了分析,但是按照质量守恒定律可知,硅酸二钙与二氧化碳反应还应生成含硅的碳化产物,而XRD结果中未见任何晶态的含硅矿物(除了未碳化的硅酸二钙),说明碳化后含硅产物是以非晶态的形式存在,这也是大多数研究未涉及含硅碳化产物结构表征的原因.FTIR和固体魔角核磁共振硅谱(29Si MAS NMR)是表征硅酸盐结构的常用方法[13].尽管29Si MAS NMR能更准确地表征硅酸盐的结构,但是由于29Si MAS NMR测试需要很长时间,而FTIR测试时间很短,且文献[13]研究了29Si MAS NMR的化学位移与FTIR波数在表征硅酸钙矿物时的关系,结果表明两者之间具有很好的线性关系,显然采取FTIR测试方法具有更好的可行性.鉴于此,本研究采用FTIR对各样品的硅酸盐结构进行表征.
各样品的FTIR图谱如图3所示.从图3(a)可以看出:硅酸二钙中硅氧键的不对称伸缩振动峰(υ3)出现在909cm-1处,表明碳化前硅酸二钙属于岛状硅酸盐结构,桥氧数为0;碳化24h后硅氧键的不对称伸缩振动峰υ3向更高波数迁移(1085cm-1,与桥氧数为4的硅酸盐结构Q4对应),表明碳化生成了高度聚合的二氧化硅凝胶;同时,碳化后硅氧键的面外弯曲振动峰(υ4)也大大减弱(514cm-1);此外,碳化后在709,876,1426cm-1处出现了新的吸收峰,分别对应方解石晶体中碳酸根的面内弯曲振动(υ2)、面外弯曲振动(υ4)和不对称伸缩振动(υ3)[12-13].从图3(b),(c)可以看出:在内掺NaCl的条件下,硅氧键的不对称伸缩振动峰υ3未发生偏移(均在1085cm-1),说明NaCl不影响碳化产物二氧化硅的聚合度,钠离子不能与二氧化硅凝胶形成化学键;此外,碳酸根的面内弯曲振动峰υ2和不对称伸缩振动峰υ3分别从709,1426cm-1偏移到748,1475cm-1,与球霰石晶体中碳酸根的振动峰一致[12,14].以上分析表明,NaCl会影响碳酸钙晶体的结构,但不影响二氧化硅凝胶的聚合度.

图3 各样品的FTIR图谱Fig.3 FTIR spectra of samples
2.4 SEM及EDS分析
XRD,TG-DTA和FTIR分析结果均表明NaCl对β-C2S矿物碳化生成的碳酸钙结构有显著影响.β-C2S矿物碳化生成的碳酸钙典型形貌如图4所示.从图4可以看出:纯β-C2S矿物碳化后生成的方解石晶体为不定形结构;在内掺NaCl的条件下,其碳化生成的球霰石为麻团状,直径大约为5~10μm.
对图4中的A,B两点进行能谱分析,结果见图5.从图5可以看出:β-C2S矿物碳化后生成的球霰石晶体中,钙、碳和氧元素的原子比大约为1∶1∶3;样品N-5-24h的碳化产物球霰石晶体中还含有钠元素,说明碳化后有部分钠固化在生成的球霰石晶体之中.

图4 样品的SEM照片Fig.4 SEM images of samples
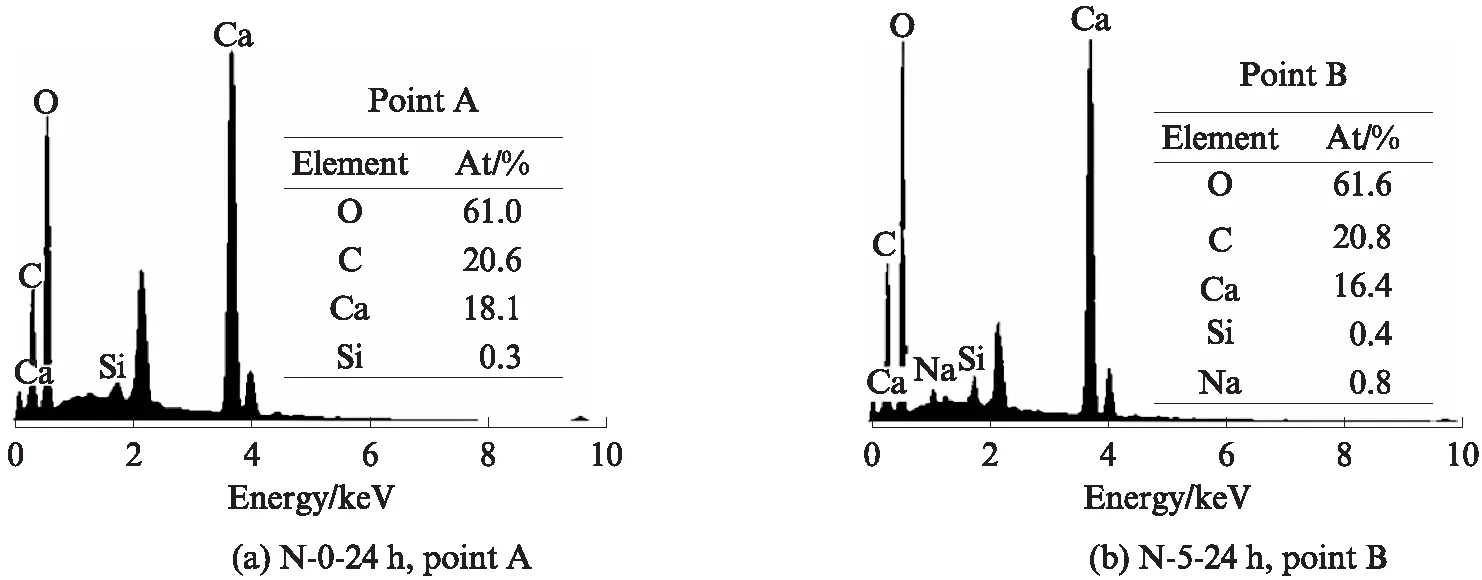
图5 样品的EDS图谱Fig.5 EDS spectra of samples
2.5 可溶碱固化机理分析
XRD定量分析结果(表1)表明,碳化后NaCl晶体含量大大降低.为了进一步验证以上结果,将各样品制成相同质量浓度的悬浮液,并测试其电导率.溶液的电导率能够在一定程度上反映可溶性离子浓度的大小.样品N-0(纯β-C2S矿物),N-5和N-10在碳化前后的电导率如图6所示.从图6可以看出:碳化前,随着NaCl掺量的提高,样品悬浮液的电导率大大提高,这是因为碳化前NaCl是可溶的,因此NaCl掺量越大,悬浮液电导率越大.碳化后,悬浮液的电导率大大降低,说明碳化后可溶性钠离子含量大大降低,但仍有一些可溶性钠离子.
众所周知,碳酸钙包括方解石、文石和球霰石3种晶体结构.其中,方解石晶体是最稳定的晶体结构,而文石和球霰石不稳定,会转化为方解石.球霰石是最不稳定的晶体结构,在自然条件下就会转化为方解石晶体.但是,以上分析均表明在钠离子存在的条件下,硅酸二钙矿物碳化生成的碳酸钙主要以稳定的球霰石晶体存在.这可能是碱金属离子的掺杂效应使球霰石晶体的结构更加稳定,从而阻止了球霰石晶体向方解石晶体的转变[14].
3 结论
(1)钠离子影响硅酸二钙矿物碳化产物的结构,但不影响其碳化程度.
(2)在钠离子存在的条件下,硅酸二钙矿物碳化生成的碳酸钙以球霰石晶体和方解石晶体的形式存在,且以球霰石为主,非晶态二氧化硅凝胶的聚合度不变.
(3)碳化反应后可溶性钠离子含量大大降低,钠离子可以被固化在球霰石晶体里,这也是球霰石晶体稳定存在的主要原因.
