Hitting a moving target: inhibition of the nuclear export receptor XPO1/CRM1 as a therapeutic approach in cancer
2018-12-19MariaSendinoMirenJosuOmaetxebarriaJoseAntonioRodrguez
Maria Sendino, Miren Josu Omaetxebarria, Jose Antonio Rodríguez
1Department of Genetics, Physical Anthropology and Animal Physiology, University of the Basque Country (UPV/EHU), Leioa 48940, Spain.
2Department of Biochemistry and Molecular Biology, University of the Basque Country (UPV/EHU), Leioa 48940, Spain.
Abstract Cellular homeostasis crucially relies on the correct nucleocytoplasmic distribution of a vast number of proteins and RNA molecules, which are shuttled in and out of the nucleus by specialized transport receptors. The nuclear export receptor XPO1, also called CRM1, mediates the translocation of hundreds of proteins and several classes of RNA to the cytoplasm,and thus regulates critical signaling pathways and cellular functions. The normal function of XPO1 appears to be often disrupted in malignant cells due to gene mutations or, most commonly, aberrant overexpression. Due to its important physiological roles and its frequent alteration in human tumors, XPO1 is a promising target for cancer therapy. XPO1 inhibitors have undergone extensive testing as therapeutic agents in preclinical models of cancer, with promising results.One of these inhibitors, Selinexor, is currently being evaluated in multiple clinical trials of different types of solid tumors and hematological malignancies. Here, we review several key aspects of XPO1 function, as well as the mechanisms that may lead to its alteration in cancer, and provide an update on the status of XPO1 inhibitors being developed as drugs for cancer therapy, including the definitive results of the first clinical trials with Selinexor that have been recently published.
Keywords: XPO1, CRM1, nucleocytoplasmic transport, Selinexor
INTRODUCTION
In 1997, a 120 kDa protein called CRM1, known to function as a chromosome region maintenance fac-tor in yeast, was identified as the first receptor for the nuclear export of proteins, and it was consequently renamed exportin 1 (XPO1)[1-4]. In these initials reports, XPO1/CRM1 (hereafter referred to as XPO1) was found to be the cellular target for a potent inhibitor of nuclear export termed leptomycin B (LMB), and to bind short amino acid sequences (so-called nuclear export signals or NESs) in proteins that were actively exported from the nucleus. Over the last two decades, many aspects of XPO1 physiopathology have been elucidated. Thus, XPO1 has been shown to mediate the nuclear export of not only hundreds of cellular and viral proteins, but also of different types of RNA molecules[5,6]. In fact, crucial signaling pathways, such as the NF-κB pathway, and essential cellular processes, such as cell cycle progression, have been shown to involve XPO1-dependent nuclear export steps[7]. In addition, export-independent functions of XPO1 in mitosis have also been identified[8]. The normal function of XPO1 appears to be often disrupted in malignant cells. Thus, overexpression of the XPO1 mRNA or protein has been frequently reported in a variety of tumor types and recurrent XPO1 gene mutations have been detected in certain hematological malignancies, suggesting that XPO1 may represent a therapeutic target in cancer[9,10]. Importantly, the results of multiple cellular, biochemical and structural analyses have led to a detailed mechanistic understanding of XPO1 function[11-13], paving the way for the development of clinically useful inhibitors of XPO1. Several compounds targeting XPO1 have been extensively tested in preclinical studies, and one of them, Selinexor,is now undergoing clinical trials, with promising results in patients with different types of cancer.
THE PHYSIOLOGICAL ROLES OF XPO1
An overview of nucleocytoplasmic transport of proteins
In eukaryotic cells, the nuclear envelope establishes a physical separation between the two major cellular compartments: the nucleus and the cytoplasm. Cellular homeostasis requires continuous communication between these compartments through the bidirectional trafficking of molecules. This trafficking may occur by diffusion in the case of small molecules, or by budding of nuclear envelope-derived vesicles for a minority of specific proteins[14]. However, the vast majority of proteins can only enter and exit the nucleus through proteinaceous channels embedded in the nuclear envelope termed nuclear pore complexes(NPCs)[15,16]. For most proteins, nucleocytoplasmic transport is an active, energy-dependent process that requires a specialized transport machinery with three crucial components: (1) the NPCs; (2) a family of soluble transport receptors (karyopherins) that recognize and bind specific transport signals in the cargo proteins; (3) a gradient of the small GTPase Ran (bound to either GTP or GDP) across the nuclear envelope, which confers directionality to the transport [Figure 1A][17-19].
NPCs, recently reviewed by Knockenhauer and Schwartz[15]and Pemberton and Paschal[17], are very large complexes (over 120 MDa in size) formed by the assembly of several copies of each of approximately 30 different proteins called nucleoporins (NUPs). NPCs present a characteristic eight-fold rotational symmetry and are composed by three stacked rings inserted into the nuclear envelope, with a series offilaments emanating to the cytoplasmic side of the NPC and a basket-like structure protruding to the nucleoplasmic side[Figure 1B]. NUPs in the inner channel of the pore contain intrinsically disordered domains rich in phenyalanine-glycine (FG) repeats. These so-called FG-nucleoporins constitute a barrier that efficiently prevents proteins above a certain size from freely diffusing across the NPC. This threshold size for exclusion has long been believed to be relatively sharp (30-60 kDa), but a recent study suggests that the NPC lacks such afirm size threshold[20]. The selective barrier of the NPC can be overcome by large proteins (and even by very large nucleoprotein complexes, such as ribosomal subunits) through binding to karyopherins[21].
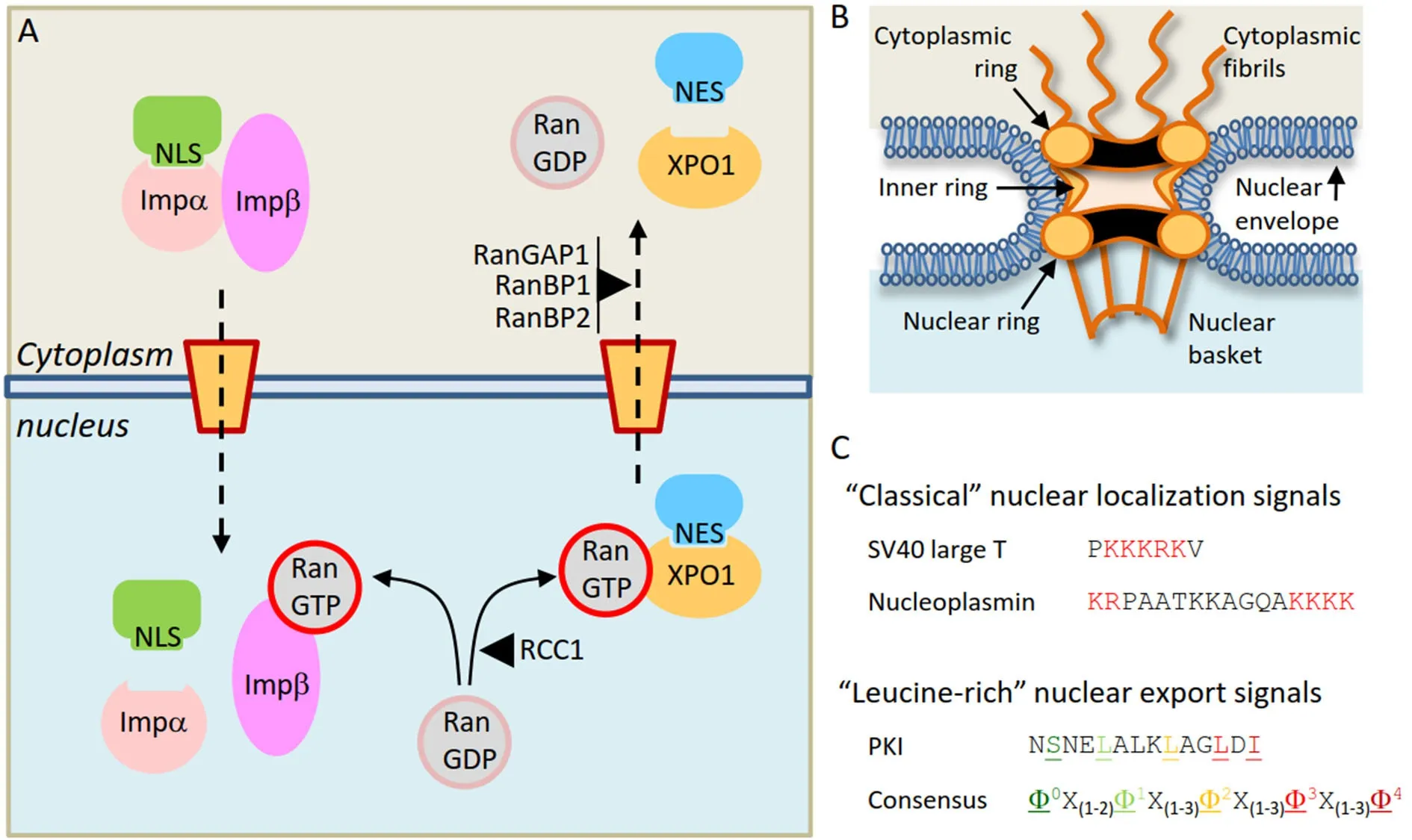
Figure 1. Receptor-mediated nucleocytoplasmic transport of proteins. A: Illustrative overview of the nuclear import of a cargo protein bearing a “classical” NLS mediated by the Importinα/Importinβ heterodimer (left) and the nuclear export of a cargo protein bearing a“leucine-rich” NES mediated by XPO1 (right); B: schematic depiction of the nuclear pore complex, illustrating its main structural features;C: examples of nucleocytoplasmic transport signals. The NLSs of SV40 large T antigen (monopartite) and nucleoplasmin (bipartite)are shown, with the basic residues that characterize “classical” NLSs highlighted in red. Below, the NES of PKI and a general consensus sequence of “leucine-rich” NESs are shown. The characteristic hydrophobic residues (represented by φ in the consensus) are highlighted in colors and underlined. NLS: nuclear localization signal; NES: nuclear export signal
The human genome codes for approximately 20 different karyopherins[22]. While some of these receptors can mediate bidirectional transport of cargos in and out of the nucleus, most of them function exclusively as either import receptors (importins) or export receptors (exportins), such as XPO1. Karyopherins can recognize and bind specific peptide sequences in the cargo protein, which function as transport signals,and can be broadly classified as nuclear localization signals (NLSs, recognized by importins) or nuclear export signals (NESs, recognized by exportins) [Figure 1C]. The best-characterized import receptor is the Importinα/Importinβ heterodimer, which mediates nuclear import of cargos bearing a “classical” NLS (a peptide sequence characterized by the presence of one or two clusters of basic residues)[23]. XPO1, the first nuclear export receptor to be identified, is also the best-characterized exportin. XPO1 mediates export of proteins bearing “leucine-rich” NESs, short peptides with a characteristic spacing of hydrophobic residues[Figure 1C]. Of note, some proteins possess both an NLS and an NES and can undergo cyclic shuttling between the nucleus and the cytoplasm[24]. Importinα/β and XPO1 mediate the nucleocytoplasmic transport of hundreds of different proteins. Other karyopherins, which are less well characterized, seem to have a more limited repertoire of cargos, and the transport signals that may mediate their binding remain, in most cases, yet to be identified.
Binding and release of a protein in the nucleus or the cytoplasm establishes the direction of its transport,and the key factor that regulates cargo binding and release by karyopherins is the small GTPase Ran,which can be bound to either GDP (RanGDP) or GTP (RanGTP)[17-19]. There is a RanGDP/RanGTP gradient across the nuclear envelope. This gradient (a high concentration of RanGDP in the cytoplasm and a high concentration of RanGTP in the nucleus) is maintained by the Ran cofactors RanGAP1 (a cytoplasmic GTPase activating protein) and RCC1 (a chromatin-bound nucleotide exchange factor). RanGTP promotes disassembly of the Importin/cargo complexes, leading to release of import cargos in the nucleus. Conversely, RanGTP stabilizes the interaction between XPO1 and export cargos in the nucleus by forming a trimeric XPO1/RanGTP/cargo complex. This complex is disassembled upon GTP hydrolysis in the cytoplasmic side of the NPC, leading to release of the export cargo in the cytoplasm. Thus, by regulating receptor/cargo interactions, the RanGTP/RanGDP gradient determines the directionality of nucleocytoplasmic transport.In fact, it has been shown that, by artificially raising the concentration of RanGTP in the cytoplasm, the direction of the transport can be inverted[25].
Beyond the basic transport machinery described above, multiple additional mechanisms may contribute to regulate the nucleocytoplasmic distribution of a given protein in a dynamic andfinely-tuned manner.These mechanisms include post-translational modifications, such as phosphorylation[26,27]or ubiquitination(reviewed by Rodríguez[28]), as well as masking/unmasking of the transport signals by homo/heterodimerization[29,30].
XPO1-mediated protein nuclear export: cargos, mechanisms and signals
XPO1 has a wide repertoire of cargos, including not only cellular proteins, but also viral proteins expressed in infected cells (reviewed by Ding et al.[31]).
Identification of XPO1 cargos has been greatly facilitated by the use of LMB as an inhibitor. In cellular experiments, cytoplasmic XPO1 cargos often relocate to the nucleus in the presence of LMB. This experimental approach cannot be used to demonstrate XPO1-mediated export of proteins that are constitutively located to the nucleus. An alternative approach in this case could be ectopic overexpression of the receptor,which promotes export of NES-containing nuclear cargos to the cytoplasm[32]. Besides LMB-based experiments, the identification, validation and characterization of XPO1 cargos often involve biochemical analyses to demonstrate RanGTP-dependent binding, as well as mutagenesis to map the NES. In over 15 years of research, hundreds of individual proteins were studied using these approaches and around 200 bona-fide XPO1-exported cargos were identified[33]. More recently, the introduction of tandem mass spectrometry(MS/MS)-based high throughput analyses has expanded the repertoire of potential XPO1 cargos (the socalled “XPO1 exportome”) to above 1000 cellular proteins[34], although many of them still need to be further validated and their NESs identified.
The search for novel cargos is still on-going, and continues to provide further insight into the physiological relevance of XPO1. For example, it has been recently found that the NES-containing protein POST and the ubiquitin-binding protein UBIN form a complex that mediates XPO1-dependent nuclear export of polyubiquitinated proteins[35], a process that seems to be exacerbated in cancer cells treated with the proteasome inhibitor bortezomib[36]. Thesefindings reveal a novel role for XPO1 in nuclear protein homeostasis that might also have important implications for cancer therapy.
From a mechanistic point of view, XPO1-mediated nuclear export consists essentially in the binding of an NES-containing protein in the nucleus and its release in the cytoplasm. The XPO1/NES interaction has low affinity, and needs to be stabilized by the cooperative binding of nuclear RanGTP, facilitated by the cofactor RanBP3[37-39]. Structural and biochemical studies carried out over the last decade have contributed to dissecting the series of molecular events that underlie the cycle of assembly and disassembly of the XPO1/RanGTP/NES complex (reviewed by Koyama and Matsuura[11], Fung and Chook[12]and Monecke et al.[13]).As schematically illustrated in Figure 2A, XPO1 is a ring-shaped protein with a concave inner surface and a convex outer surface. RanGTP binds to the inner surface, and NESs dock into a hydrophobic groove in the outer surface of the receptor. The open/close state of the NES binding groove is allosterically regulated by conformational rearrangements of two additional XPO1 structural elements, termed the H9 loop and the C-terminal extension, which play a crucial role in the cycle of NES binding and release.
As illustrated in Figure 1C, “leucine-rich” NESs conform to a loose consensus sequence with a characteristic spacing of hydrophobic residues[40,41]. Hundreds of different amino acid sequences have been experimentally validated as bona-fide NESs that bind the receptor with different affinity, and may be exported with different efficiency[33,42,43]. This high variability can be explained by the recentfinding that NESs with differ-ent backbone conformations can bind the receptor, and that not all export signals occupy the XPO1 NES-binding groove to the same extent[44].
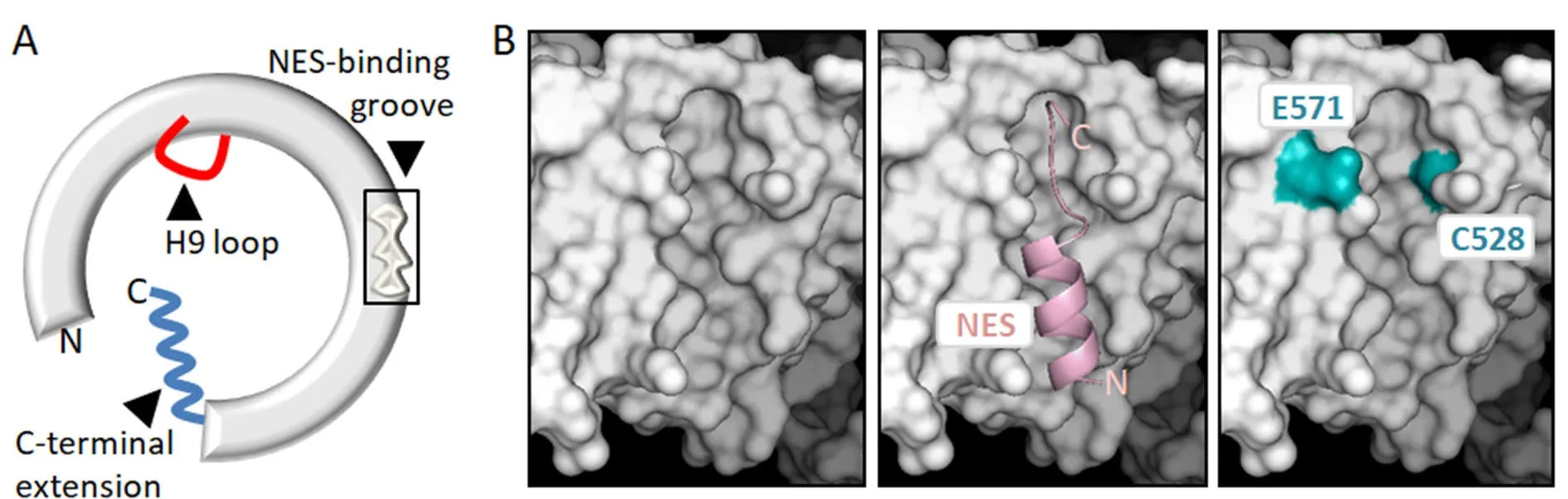
Figure 2. Structural features of XPO1 related to its nuclear export function, its role in cancer and its potential as a therapeutic target. A:Schematic representation of XPO1 protein illustrating its general ring-shaped conformation, and showing the three structural motifs that are crucial for its function as a nuclear export receptor: the NES-binding groove, the H9 loop and the C-terminal extension; B: detailed views of the NES-binding groove on the molecular surface of XPO1. The UCSF Chimera package[206] and XPO1 structure 3GJX[207] were used to generate the images. The left panel shows the empty groove, the middle panel shows a “leucine-rich” NES peptide (pink) bound to the groove. The right panel shows residues E571 and C528 highlighted in blue. E571 is a mutational hotspot in several hematological malignancies. C528 is the residue targeted by XPO1-inhibiting drugs, such as LMB or Selinexor. These compounds attach covalently to C528 and physically occupy the groove, blocking NES binding. NES: nuclear export signal; LMB: leptomycin B
“Leucine-rich” NESs dock into the groove and engage in predominantly hydrophobic interactions with several XPO1 residues. Two non-hydrophobic amino acids (C528 and E571) located in or near the NES-binding groove [Figure 2B] are of particular interest regarding the targeting of XPO1 and its potential role in cancer. On one hand, the amino acid E571 is recurrently mutated in certain hematological malignancies (see below), suggesting that mutation of this particular residue can be a driver event in some types of cancer. On the other hand, C528 is the crucial target for the effect of LMB and more clinically relevant XPO1 inhibitors, which covalently bind to this residue and block NES binding by physically occupying the groove. In fact, experimental mutation of C528 renders cells resistant to these inhibitors[45].
Role of XPO1 in RNA nuclear export
Following transcription in the nucleus, active export to the cytoplasm is an essential step during the biogenesis of many classes of RNA, and/or a critical requirement for their function (recently reviewed by Williams et al.[46]). Thus, messenger RNAs (mRNAs) need to be exported to undergo translation into proteins, while ribosomal RNAs (rRNAs), transfer RNAs (tRNAs), small nuclear RNAs (snRNAs), microRNAs(miRNAs) and long non-coding RNAs (lncRNAs) need to be transported to the cytoplasm in order to be processed or to carry out their cellular activities. Nuclear export of RNA is a tightly regulated process that involves the coordinated function of many different factors, including a large array of RNA-binding adaptor proteins as well as dedicated export receptors[6]. XPO1 plays a pervasive role in this process, mediating the nuclear export of several different classes of RNA.
XPO1 plays a prominent role in the export of 40S and 60S ribosomal subunits, containing rRNA, to the cytoplasm, which is a necessary step for theirfinal maturation. The NES-containing protein Nmd3 functions as the adaptor for 60S subunit export, while the adaptor involved in the export of the 40S subunit remains to be identified[47].
In contrast to rRNAs, the vast majority of cellular mRNAs are exported to the cytoplasm by a receptor unrelated to karyopherins, called NXF1, but XPO1 mediates nuclear export of a subset of mRNAs[48]. Since XPO1 does not bind mRNA directly, NES-containing RNA-binding proteins that act as adaptors to bridge the interaction between XPO1 and mRNA are essential in this process. These adaptors include NXF3[49],and the HuR/APRIL/pp32[50]or the eIF4E/LRPPRC[51]complexes. Interestingly, several mRNAs exported by XPO1 code for proteins that are involved in tumorigenesis-related processes, such as invasion and metastasis[46].
Finally, some RNA species with an important role in the regulation of gene expression (snRNA, lncRNA and miRNA) can also be exported by XPO1[52-54]. For example, although the major exporter of miRNAs is not XPO1 but another exportin called XPO5, XPO1 mediated-export plays a role in the export and biogenesis of specific subsets of miRNAs[55,56]. Intriguingly, XPO1 has also been reported to mediate the nuclear import of mature miRNAs[57].
Nuclear export-independent role of XPO1 as a key regulator of mitosis
Besides mediating the export of proteins and RNA to the cytoplasm, XPO1 also plays a role in processes that do not directly involve nuclear export, such as intranuclear trafficking of small nucleolar RNAs (snoRNAs)from Cajal bodies to the nucleolus[58]. A particularly relevant aspect of cell physiology where XPO1 carries out export-independent functions is mitosis[8]. This role of XPO1 has been reviewed by Forbes et al.[59].
In eukaryotic cells undergoing open mitosis, the breakdown of the nuclear envelope at the onset of prometaphase dramatically disrupts the nucleocytoplasmic compartmentalization. With no physical separation between nucleus and cytoplasm, the nuclear transport machinery, including certain transport receptors, NUPs and the Ran GTPase, is “repurposed” to carry out transport-independent mitotic functions,such as regulating the assembly of the mitotic spindle[59]. In this context, XPO1 has been shown to function as a “mitotic effector” of Ran, mediating RanGTP-dependent targeting of key mitotic proteins to specific spindle structures, such as the centrosomes or the kinetochore. Thus, the NES-containing protein pericentrin, a crucial scaffold for microtubule nucleation at the spindle poles, is recruited to the centrosomes by XPO1 in a RanGTP-containing trimeric complex that resembles the nuclear export complexes described above[60]. On the other hand, the stable microtubule-kinetochore interactions necessary for proper chromosome segregation appear to require XPO1-mediated recruitment of a protein complex containing RanGTP,RanGAP1 and the nucleoporin RanBP2 to the kinetochores[8].
The mitotic functions of XPO1, like its nuclear export activity, seem to be the subject of careful regulation through mechanisms that include phosphorylation[61]and competition with importins[62].
In summary, although its primary role may be in protein nuclear export, XPO1 is a multifaceted protein with roles in other processes. This functional complexity should be taken into account when interpreting the results of XPO1 inhibition studies.
PATHOLOGICAL ALTERATION OF XPO1 IN CANCER
Altered nucleocytoplasmic localization of proteins in cancer
Normal cell function relies on the correct subcellular distribution of thousands of proteins. The presence of a critical protein in the wrong cellular compartment may have severe pathological consequences. For example, aberrant cytoplasmic localization of a physiologically nuclear tumor suppressor protein may render this protein inactive, and thus contribute to tumorigenesis. In fact, mislocalization of cancer-related proteins, including the products of prominent oncogenes and tumor suppressor genes, has been often demonstrated in human tumors[63,64-66].
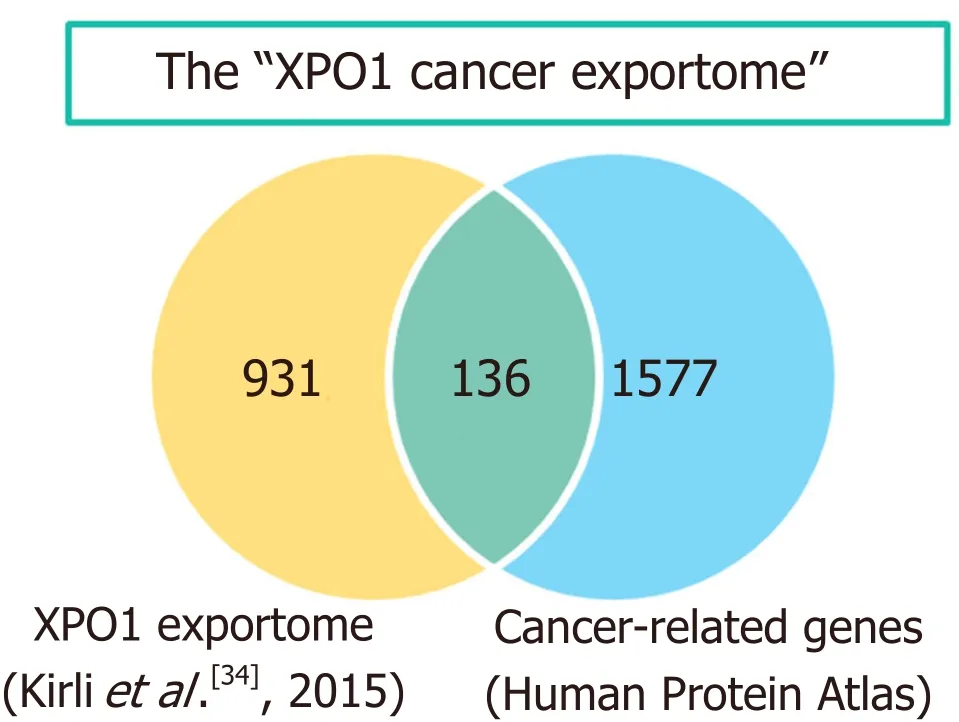
Figure 3. The “XPO1 cancer exportome”. The Venn diagram shows the overlap between the list of potential XPO1 cargos identified in HeLa cells[34] and the group of “cancer related genes” defined in the Human Protein Atlas (v.18). The 136 overlapping proteins represent what could be referred to as the “XPO1 cancer exportome”. The diagram was created using the jvenn web tool[208]
Nucleocytoplasmic localization of proteins can be disrupted by different mechanisms in cancer cells. On one hand, the trafficking of a specific protein can be altered by mutations that either interfere with the activity of its transport signals (NLSs or NESs) or that create a novel signal in the mutant protein. For example, aberrant localization of tumor suppressors BRCA2[67]and PALB2[68]to the cytoplasm can result from mutations that unmask normally hidden NESs, whereas cytoplasmic mislocalization of certain NPM1 mutants is the result of a frameshift mutation that creates a novel strong NES, not present in the wild-type protein[69].
On the other hand, a general defect in the nucleocytoplasmic localization of proteins (and RNA) may arise in tumor cells, if elements of the transport machinery themselves are genetically altered or aberrantly expressed[70]. Examples of genetic alterations targeting the nuclear transport machinery include chromosome rearrangements involving nucleoporin genes (e.g., NUP98 and NUP214) in hematologic malignancies[71].The abnormal fusion proteins resulting from these translocations have been reported to disrupt XPO1-mediated export[72,73]. Examples of nuclear transport factors abnormally expressed in tumors include the nuclear import receptors Importin β (see Dickmanns et al.[64]and references therein) and Importinα1 (see Christiansen and Dyrskjøt[74], and references therein).
In the case of XPO1, both aberrant expression and genetic alterations have been detected in different types of cancer, as detailed below. The abnormal XPO1 function that may result from these alterations would, in turn, hinder the normal nucleocytoplasmic localization of hundreds of XPO1 cargo proteins. In the context of the present review, those XPO1 cargos with a known role in the development of human tumors are of particular interest. In this regard, we note that the extended list of potential XPO1 cargos identified in HeLa cells by a recent high throughput analysis[34]includes 136 members of the protein class “cancer-related genes” registered in the Human Protein Atlas initiative (https://www.proteinatlas.org/) [Figure 3]. The set of cancer-related proteins exported by XPO1 (which could be referred to as the “XPO1 cancer exportome”)includes prominent tumor suppressors, such as p53[75]and BRCA1[76], as well as protooncogenes, such as cabl[77]. A more extensive account of cancer-related proteins that undergo XPO1-mediated nuclear export can be found in previous reviews[10,65,66,78,79].
Altered XPO1 expression in human tumors
The expression level of XPO1 at either the mRNA or protein level has been analyzed in many different cancer types. As summarized in Table 1, XPO1 is frequently overexpressed in tumor samples with respect to the corresponding normal tissue samples[80-99]. In fact, XPO1 overexpression was observed in all solid tumor types and hematologic malignances examined, with the exception of liver cancer[91].
In several of these studies, the potential prognostic significance of XPO1 expression has been evaluated.Higher XPO1 expression was associated with poorer patient prognosis in patients with ovarian tumors[90],pancreatic tumors[94], esophageal tumors[92], gliomas[84,85], thymic epithelial tumors[89], and breast tumors[96].In contrast, high XPO1 expression was related to better prognosis in osteosarcoma patients[98]. Finally, contradictoryfindings on the prognostic value of XPO1 expression in gastric cancer have been reported[87,88].
The molecular mechanisms responsible for XPO1 overexpression in cancer cells are still poorly characterized. Copy number gains at chromosomal region 2p, affecting the XPO1 locus, have been found to correlate with high XPO1 mRNA expression in lymphomas[100]and chronic lymphocytic leukemia (CLL)[101].In addition, XPO1 transcription has been reported to be regulated by cMyc and p53[102,103], two proteins that are frequently altered in cancer. Conceivably, disruption of this regulation may contribute to aberrant XPO1 expression in some tumors, although further studies are required to test this possibility.
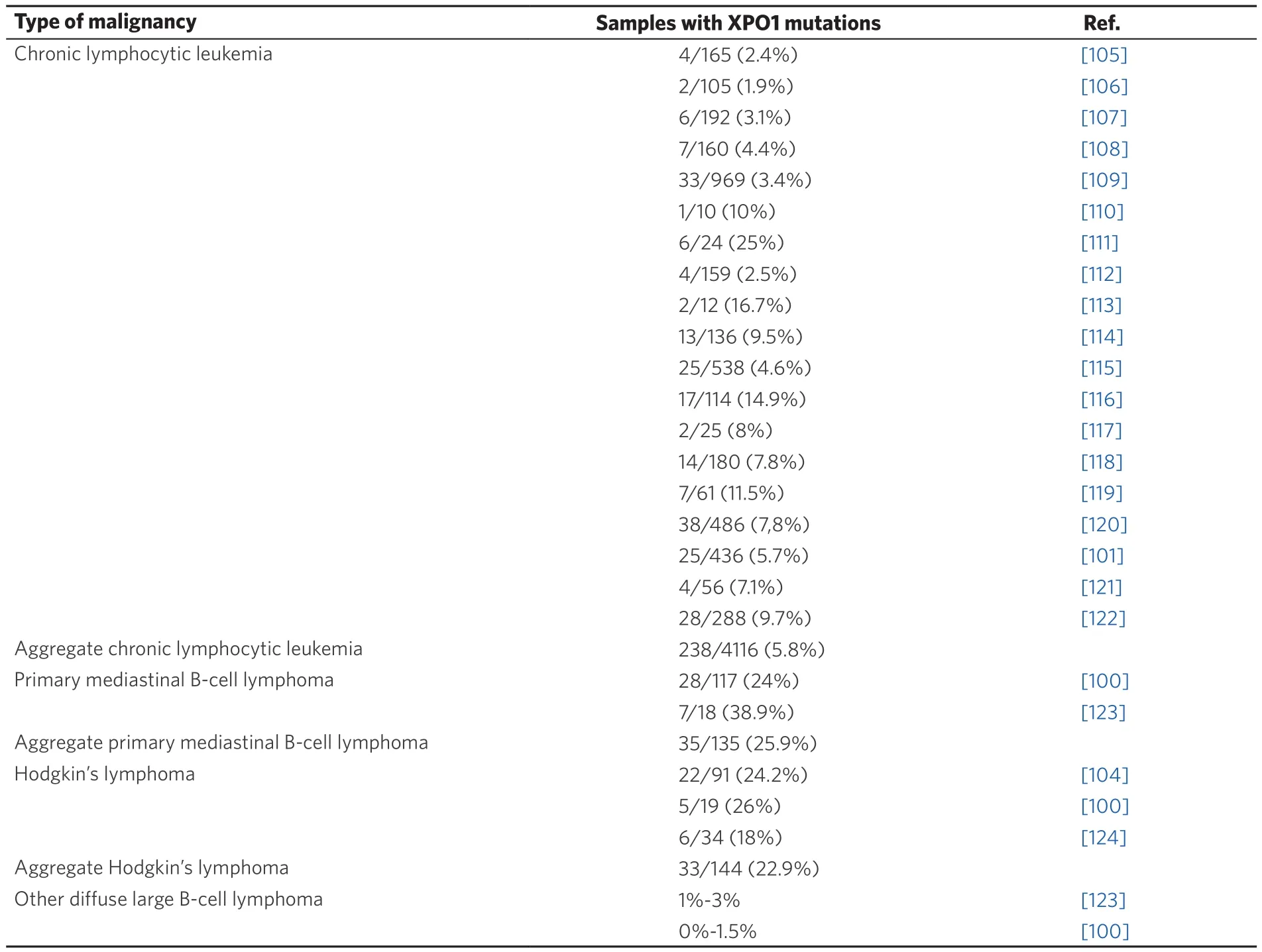
Table 2. Recurrent XPO1 mutations in hematological malignancies. More than 90% of the reported mutations are missense changes affecting XPO1 “hotspot” residue E571
A recurrent XPO1 gene mutation in hematological malignancies
Missense mutations of the glutamic residue E571 (mostly E571K) have been detected in around 25% of patients with two specific types of hematological malignancies: primary mediastinal B-cell lymphoma(PMBL)[100], and classical Hodgkin’s lymphoma (HL)[104]. The E571 “hotspot” mutations in XPO1 were first detected by whole genome sequencing analysis of CLL samples[105]. A large number of targeted studies[100,101,104-124][Table 2] have subsequently confirmed the presence of E571 missense mutations in around 5%of CLL patients.
In CLL, the presence of XPO1 mutations is often associated with unmutated IGHV status[105,109,120], but does not seem to be a marker of poor patient prognosis[120]. Similarly, HL patients with XPO1 mutations do not appear to have a worse prognosis than patients with wild type XPO1[104]. In contrast, a shorter progressionfree survival was reported for PMBL patients bearing XPO1 mutations[100]. Interestingly, it has been suggested that XPO1 mutations could represent useful biomarkers to evaluate minimal residual disease in HL and PMBL[125].
Isolated instances of mutant XPO1 have been reported in esophageal[126]and thyroid cancer[127], but XPO1 genetic alterations seem to be a very rare event in solid tumors.
It is still unclear why XPO1 E571 mutations are particularly common in certain types of cancer, and why they may have different prognostic significance in different types of hematological malignancies. In fact,the molecular mechanisms that may be responsible for the pathogenic effect of XPO1 mutations remain to be established. Consistent with the location of the mutational “hotspot” proximal to the NES-binding site[Figure 2B], it has been reported that the E571K mutation subtly increases the affinity of the receptor for some NESs with a negatively charged carboxy-terminal end[32]. Conceivably, this could lead to altered export of one or more cargos, whose mislocalization might in turn contribute to tumorigenesis.
Given its frequent alteration in human tumors, and its crucial cellular roles described above, XPO1 has long been regarded as a potentially relevant target in cancer therapy.
XPO1 INHIBITION IN CANCER THERAPY
Development and preclinical evaluation of selective inhibitors of nuclear export
Even before XPO1 was identified as its cellular target, LMB (also called elactocin) had been found to possess antitumor activity, and it had been tested in a clinical trial[128].
LMB was found to have severe toxicities when administered to patients, precluding its development as a clinically useful drug[128]. Nevertheless, the availability of this potent and specific inhibitor made it possible to carry out proof-of-concept experiments testing the effect of XPO1 inhibition in different tumor settings.As an illustrative example, we will brie fl y describe some early data regarding the effect of LMB treatment in chronic myelogenous leukemia (CML) cells expressing the BCR-ABL oncoprotein. Shortly after the identification of XPO1 as a nuclear export receptor, the c-ABL kinase was identified as one of its cargos,bearing a C-terminal NES that is also present in the BCR-ABL fusion protein[77]. At that time, treatment of BCR-ABL-positive CML patients was undergoing a dramatic improvement with the introduction of the kinase inhibitor imatinib[129,130]. In this context, it was reported that the sequential combination of imatinib plus LMB led to the nuclear entrapment of BCR-ABL, which selectively induced apoptosis of CML cells[131].Furthermore, subsequent experiments showed that the combination with LMB could overcome imatinib resistance due to BCR-ABL amplification[132].
These and other encouragingfindings in different tumor types (reviewed by Turner and Sullivan[7]) suggested that XPO1 inhibition might represent a valid strategy for cancer treatment, fostering the search for other XPO1 inhibitors. Over the next years, several natural and synthetic inhibitors of XPO1 were reported(reviewed by Tan et al.[10]and Senapedis et al.[133]). Similar to LMB, these compounds bind covalently to XPO1 residue C528, and occupy the NES-binding groove, blocking access to NESs. However, unlike LMB,some of these novel inhibitors, such as CBS9106 or S109, bind to XPO1 in a reversible manner, which was associated to less severe toxicity in preclinical in vivo models[84,134]. Studies with these compounds further validated XPO1 inhibition as a relevant strategy for cancer treatment. For example, blocking nuclear export of topoisomerase II with the XPO1 inhibitor Ratjadone C was found to sensitize multiple myeloma(MM) cells to doxorubicin and etoposide[135].
While most XPO1 inhibitors have only been tested in vitro or in mouse xenograft, there is a series of compounds, termed selective inhibitors of nuclear export (SINEs) that are undergoing development as potential anticancer drugs, and some of these compounds are already being evaluated in clinical trials[133].
SINEs were developed in 2012 using structure-assisted relationship methodology combined with a novel computational approach termed consensus-inducedfit docking[136,137], a strategy that relied crucially on the recently solved structures of NES-bound XPO1. The “ first-generation” series of SINEs included a relative large number of slowly reversible XPO1 inhibitors, such as KPT-127, KPT-185, KPT-205, KPT-227, KPT-249, KPT-251, KPT-276 and KPT-330 (Selinexor). As summarized in Tables 3 and 4, SINE compounds have been extensively tested in preclinical models of many different hematological malignancies[80,82,83,100,101,137-157]and solid tumors[89,95,97,158-188]. In these models, SINEs have demonstrated potent in vitro and in vivo activity against cancer cells (including growth inhibition, induction of apoptosis, and cell cycle arrest), with only minor toxic effects on normal cells. Importantly, several SINEs (most prominently Selinexor) have been shown to increase the sensitivity of cancer cells to currently used drugs, such as doxorubicin or the proteasome inhibitors bortezomib and carfilzomib, and also to synergize with other targeted therapeutic agents,such as ibrutinib (an inhibitor of Bruton tyrosine kinase) or linsitinib (an inhibitor of insulin-like growth factor receptor-1). A more extensive and detailed discussion of the preclinical results obtained with SINEs in specific tumor settings can be found in recent reviews[9,10,79,133,189].
In general terms, the anticancer effect of XPO1 inhibition is thought to rely on the relocation of mislocalized XPO1 cargos with tumor-suppressive and growth-regulatory functions (e.g., p53) to the nucleus, where they carry out their normal activity. In our opinion, this may be an overly simplistic view. Given the large number of potential XPO1 cargos with a role in cancer, the export-independent roles of XPO1, and the complex nature of the tumorigenesis process, the specific molecular and cellular mechanisms underlying the anticancer effect of SINEs may differ in different tumor settings. In this regard, as indicated in Tables 3 and 4, preclinical studies are providing important information on tumor context-specific proteins and signaling pathways that may mediate SINE activity, such as the BCR-ABL oncoprotein in CML mentioned above, or the NF-κB pathway in lung cancer[176].
Intriguingly, there is emerging evidence that, in addition to cancer, other conditions, such as demyelinating diseases[190]or viral infections[191]might be amenable to treatment with SINEs.
Evaluation of Selinexor in clinical trials
In preclinical studies, Selinexor compared favorably to other “ first-generation” SINEs in terms of the balance between potency and tolerability. Selinexor, an orally available drug, is the only compound of the series that has advanced into clinical development for human cancer.
The ClinicalTrials.gov site (https://clinicaltrials.gov/, accessed on 11 Jun 2018) registers 60 clinical studies on different tumor types with Selinexor as single agent or in combination with other drugs. In addition,there are isolated clinical trials registered for other XPO1 inhibitors, such as the “second-generation” SINE compound KPT-8602 (Eltanexor) or the non-SINE compound SL-801.
Interim data from some clinical studies with Selinexor have been reported as meeting proceedings (some of these data are reviewed by Mahipal and Malafa[192]). Here, we will limit our discussion to the results of phase I and II trials that have undergone full peer-reviewed publication as PubMed-indexed articles (summarized in Table 5).
Abdul Razak et al.[193]evaluated the safety, pharmacokinetics, pharmacodynamics, and efficacy of Selinexor in 189 patients with advanced solid tumors, testing several doses and administration schedules. The most common grade 3 or 4 toxicities in this series were thrombocytopenia, fatigue, and hyponatremia. One hundred and fifty seven patients were evaluable for response. Seven patients achieved partial or complete response, and 27 patients achieved stable disease for ≥ 4 months. The authors concluded that Selinexor is a safe therapeutic with broad antitumor activity, and proposed a recommended phase II dose (RP2D) of 35 mg/m2with a twice-a-week dosing schedule.
Gounder et al.[194]carried out a phase I study on 54 patients with advanced soft tissue or bone sarcoma.Selinexor was administered twice per week at doses of 30 mg/m2, 50 mg/m2, or 60 mg fl at dose. The mostcommon grade 3 or 4 toxicities in this series were fatigue, thrombocytopenia, anemia, lymphopenia, and leucopenia. In 52 evaluable patients, no objective responses were seen, but 17 patients achieved stable disease for ≥ 4 months. The authors concluded that Selinexor shows preliminary evidence of anticancer activity in sarcoma, and is well tolerated at a 60 mg fl at dose.

Table 3. Summary of preclinical studies with “ first-generation” SINEs in hematological malignancies

Table 4. Summary of preclinical studies with “ first-generation” SINEs in human solid tumors
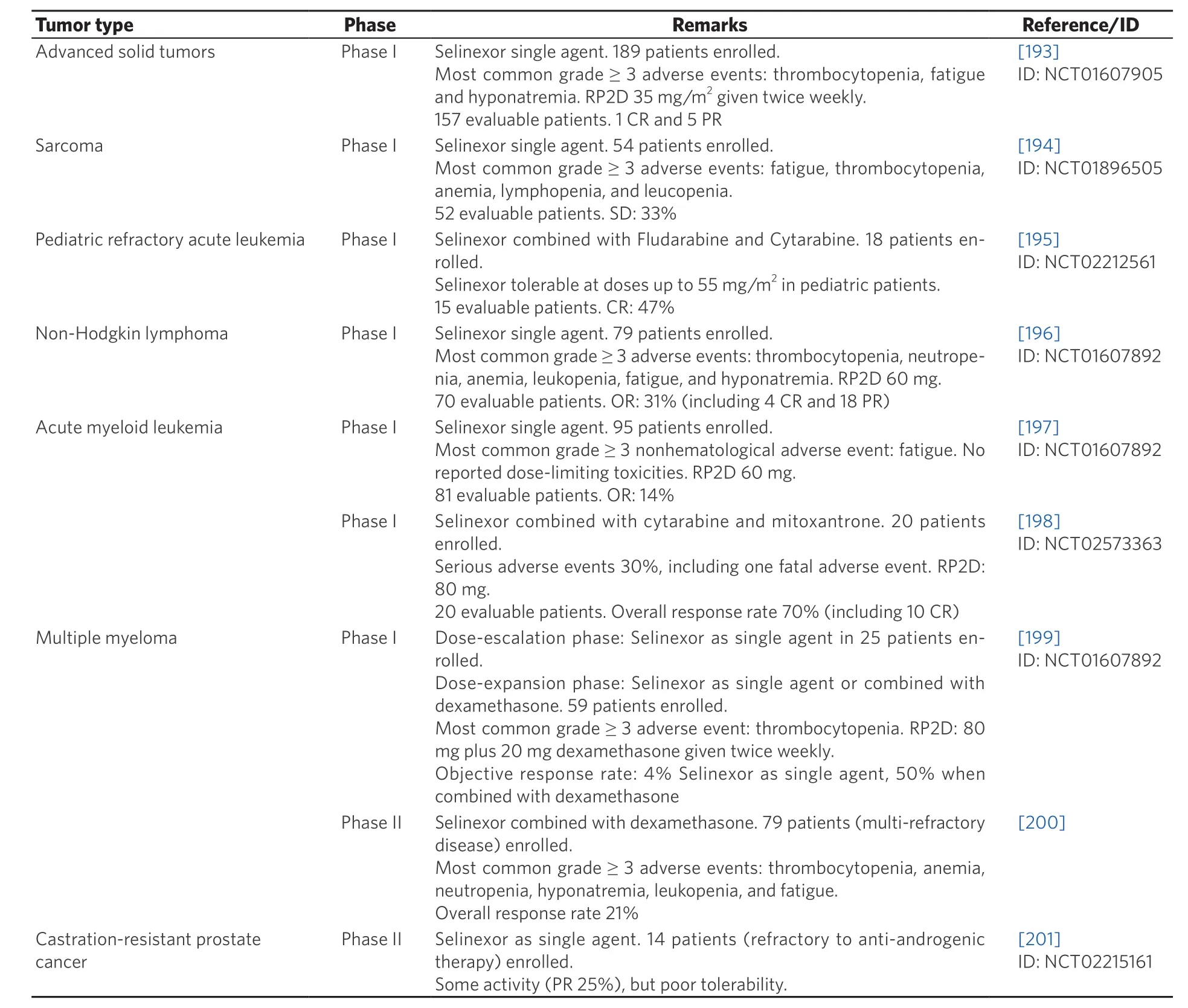
Table 5. Summary of the results of published clinical studies of Selinexor in human malignancies
Alexander et al.[195]evaluated the combination of Selinexor with fl udarabine and cytarabine in 18 pediatric patients with relapsed or refractory leukemia. Selinexor was administered twice per week at several doses between 30 mg/m2and 70 mg/m2. Dose-limiting reversible cerebellar toxicity was experienced by some patients treated with the 70 mg/m2dose. Seven of the 15 patients that were evaluable achieved complete response. The authors concluded that Selinexor combined with fl udarabine and cytarabine shows promising response rates in pediatric patients with relapsed or refractory leukemia. A 55 mg/m2dose Selinexor is tolerable in this combination.
Kuruvilla et al.[196]evaluated Selinexor in 79 patients with different subtypes of non-Hodgkin lymphoma(NHL). In the dose-expansion phase of the study, Selinexor was administered at doses of 35 mg/m2or 60 mg/m2. The most common grade 3 or 4 toxicities in this series were thrombocytopenia, neutropenia,anemia, leukopenia, fatigue, and hyponatremia. Twenty-two objective responses (4 of them complete responses) were observed in 70 evaluable patients. The authors concluded that Selinexor shows encouraging activity in NHL patients, and proposed 35 mg/m2Selinexor (60 mg fl at dose) as the RP2D.
Garzon et al.[197]carried out a phase I dose-escalation study in 95 patients with relapsed or refractory acute myeloid leukemia (AML). Several doses and administration schedules were tested. The most common grade 3 or 4 toxicities were thrombocytopenia, anemia, fatigue, and neutropenia. Objective responses were observed in 11 of the 81 evaluable patients. The authors concluded that Selinexor is a safe therapy in AML patients, and established the RP2D at 35 mg/m2(60 mg fl at dose) given twice weekly. Another phase I doseescalation trial in AML evaluated the combination of Selinexor with high-dose cytarabine and mitoxantrone in 20 patients[198]. Selinexor doses of 60 mg or 80 mg were administered. Serious toxicities, including one fatal adverse event, occurred in 30% of the patients. The overall response rate was 70%, including 10 patients achieving complete remission. The authors concluded that Selinexor combined with high-dose cytarabine and mitoxantrone is a feasible and tolerable treatment in AML patients, and proposed 80 mg Selinexor twice weekly as the RP2D in this combination.
Chen et al.[199]evaluated Selinexor in 84 patients with heavily pretreated MM (81 patients) or Waldenstrom macroglobulinemia (3 patients). Single agent Selinexor was given to 25 patients in the dose-escalation phase. In the dose-expansion phase, Selinexor was administered in combination with dexamethasone to 59 patients. The most commonly reported grade 3 or 4 toxicity in this series was thrombocytopenia. Although the efficacy of Selinexor as single agent was modest, its combination with dexamethasone resulted in a significantly increased activity, with an objective response rate of 50%. The authors proposed a RP2D of 80 mg Selinexor plus 20 mg dexamethasone given twice weekly. This treatment regimen was administered to 79 patients with multi-refractory MM in a recently reported phase II study[200]. The overall response rate(the primary endpoint of the study) was 21% and the most frequent grade 3 or 4 toxicity was thrombocytopenia. In relation to these studies, a more extensive and detailed discussion on the clinical implementation of Selinexor in MM has been recently published[189].
The last clinical study on Selinexor published to date (May 2018) is a phase II trial that evaluated its efficacy and tolerability in 14 patients with metastatic, castration-resistant, prostate cancer[201]. Selinexor was administered twice weekly at a dose of 65 mg/m2that had to be subsequently reduced to a fl at dose of 60 mg to improve tolerability. In fact, although Selinexor showed some evidence of clinical activity (reduction in prostate-specific antigen levels, and radiographic response), it was poorly tolerated in this patient population.In summary, the results of these studies show that Selinexor, as single agent or in combination with other drugs, has broad clinical activity in multiple types of solid tumors and hematological malignancies and is generally well tolerated by patients. One of the most common high-grade toxicities experienced by patients treated with Selinexor is thrombocytopenia. The mechanism underlying this adverse event has been recently elucidated. Machlus et al.[202]have shown that thrombocytopenia results from reduced maturation of megakaryocyte progenitors due to Selinexor-mediated inhibition of thrombopoietin signaling. Importantly, the severity of thrombocytopenia could be reduced by temporary interruption of Selinexor treatment and administration of thrombopoietin mimetics to patients[202].
Altogether, these clinical data support the view that inhibition of XPO1 represents a valid therapeutic strategy in cancer.
CONCLUSION AND FUTURE DIRECTIONS
Twenty years after its identification as a receptor that mediates the nuclear export of proteins, there is compelling evidence that XPO1 represents a relevant target in cancer. Further basic, preclinical and clinical in-vestigations are required to address several salient questions on the use of XPO1 inhibition as a therapeutic approach.
On one hand, novel XPO1 inhibitors with more favorable clinical properties are being developed. In this regard, a “second-generation” SINE (KPT-8602 or Eltanexor) has demonstrated improved tolerability in preclinical models[203-205]and is currently undergoing clinical evaluation.
On the other hand, it is necessary to further elucidate the mechanisms that mediate the oncogenic role of XPO1 alterations (overexpression or mutation) in different types of cancer and to better characterize the molecular and cellular mechanisms underlying the effect of XPO1 inhibitors. This basic mechanistic information, which is still rather limited, would be crucial to successfully implement XPO1-targeting drugs in the clinic, as it could help to design rational combinations with other agents, to identify subsets of patients that may benefit more from the treatment and to improve the clinical management of adverse effects.
DECLARATIONS
Acknowledgments
We thank our colleagues at the UPV/EHU Dr. Sonia Bañuelos (Dept. of Biochemistry and Molecular Biology), Dr. Gorka Prieto (Dept. of Communication Engineering) and Dr. Asier Fullaondo (Dept. of Genetics,Physical Anthropology and Animal Physiology) for their help in preparing Figures 2 and 3. Supported by grants from the Spanish Government MINECO-FEDER (SAF2014-57743-R), the Basque Country Government (IT634-13) and the University of the Basque Country (UFI11/20), as well as a fellowship from the Basque Country Government (to MS).
Authors’ contributions
Conception of the work, draft and revision of the work: Sendino M, Omaetxebarria MJ, Rodríguez JA
Availability of data and materials
Not applicable.
Financial support and sponsorship
None.
Conflicts of interest
All authors declared that there are no con fl icts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2018.