Linking tyrosine kinase inhibitor-mediated inflammation with normal epithelial cell homeostasis and tumor therapeutic responses
2018-12-19NataliaGuruleLynnHeasley
Natalia J. Gurule, Lynn E. Heasley
Department of Craniofacial Biology, University of Colorado Anschutz Medical Campus, Aurora, CO 80045, USA.
Abstract Receptor tyrosine kinases (RTKs) bearing oncogenic mutations in EGFR, ALK and ROS1 occur in a significant subset of lung adenocarcinomas. Tyrosine kinase inhibitors (TKIs) targeting tumor cells dependent on these oncogenic RTKs yield tumor shrinkage, but also a variety of adverse events. Skin toxicities, hematological deficiencies, nausea, vomiting,diarrhea, and headache are among the most common, with more acute and often fatal side effects such as liver failure and interstitial lung disease occurring less frequently. In normal epithelia, RTKs regulate tissue homeostasis. For example, EGFR maintains keratinocyte homeostasis while MET regulates processes associated with tissue remodeling.Previous studies suggest that the acneiform rash occurring in response to EGFR inhibition is a part of an inflammatory response driven by pronounced cytokine and chemokine release and recruitment of distinct immune cell populations.Mechanistically, blockade of EGFR causes a Type I interferon response within keratinocytes and in carcinoma cells driven by this RTK. This innate immune response within the tumor microenvironment (TME) involves increased antigen presentation and effector T cell recruitment that may participate in therapy response. This TKI-mediated release of inflammatory suppression represents a novel tumor cell vulnerability that may be exploited by combining TKIs with immune-oncology agents that rely on T-cell inflammation for efficacy. However, early clinical data indicate that combination therapies enhance the frequency and magnitude of the more acute adverse events, especially pneumonitis,hepatitis, and pulmonary fibrosis. Further preclinical studies to understand TKI mediated inflammation and crosstalk between normal epithelial cells, cancer cells, and the TME are necessary to improve treatment regimens for patients with RTK-driven carcinomas.
Keywords: Tyrosine kinase inhibitor, receptor tyrosine kinase, interferon, inflammation, tumor microenvironment,epithelial tissue homeostasis
ONCOGENIC RECEPTOR TYROSINE KINASES AS TARGETS FOR PRECISION MEDICINE
Malignancies of epithelial tissues account for 80 to 90 percent of all cancer cases, making carcinomas the most common histological type of cancer[1]. Activating mutations in receptor tyrosine kinases (RTKs) and their associated downstream signal pathways function as oncogene drivers in many solid tumor types. Lung adenocarcinomas (LUADs) serve as an example of a carcinoma arising from distinct pulmonary epithelial cells that harbor many diverse oncogenic RTKs including EGFR, ALK, MET, and ROS1[2]. Moreover, specific tyrosine kinase inhibitors (TKIs) are now routinely deployed as first-line therapies in patients with lung tumors presenting with these oncogenic RTKs. Examples of these TKIs include gefitinib or osimertinib for EGFR, and crizotinib or ceritinib for ALK, ROS1 and MET. Cetuximab, a monoclonal antibody against EGFR is also used to treat patients with head and neck squamous carcinoma (HNSCC), which often overexpress EGFR[3]. Thus, these oncogene targeted agents have proven efficacious for inducing tumor regression as firstline therapies, although complete responses are rare and emergence of acquired resistance is universal[4].
Although TKIs are less toxic than traditional cytotoxic drugs, their use is still associated with various adverse effects including skin toxicity, hematological deficiencies, nausea, vomiting, diarrhea, and headaches being the most common side effects. Skin toxicities are very frequent, occurring in 49%-95% of patients treated with EGFR inhibitors, and 16% of patients treated with ALK/c-MET inhibitors[5-7]. More acute and often fatal side effects such as liver toxicity and forms of interstitial lung disease (ILD) occur at a lower frequency in cancer patients treated with the TKIs gefitinib, erlotinib, and crizotinib[8,9]. ILDs such as pneumonitis and pulmonaryfibrosis occur at frequencies of < 1% and 1.6% respectively with EGFR inhibitors and ALK inhibitors[5,10].
The role of EGFR in regulating cellular proliferation, survival, and differentiation during development, tissue homeostasis, and carcinogenesis is well established. EGFR is expressed in a variety of normal epithelial tissues including skin[11]. Within the epidermis, EGFR is most prominently expressed in proliferating basal and suprabasal keratinocytes. In keratinocytes, EGFR signaling sustains proliferation and migration and delays apoptosis in suprabasal keratinocytes that are no longer attached to matrix[12-14]. In addition to normal keratinocyte dependent skin homeostasis, EGFR signaling functions in the protective response triggered by epithelial cells during wound healing or during defense against microorganisms that cause skin infections.EGFR is also highly expressed in alveolar type II epithelial cells in the lung[15,16]. The MET tyrosine kinase receptor and its ligand HGF have well characterized functions in tissue remodeling via regulating cellular processes such as proliferation, apoptosis, morphogenic differentiation, motility, invasion and angiogenesis.MET is expressed on the surface of epithelial cells in the liver, pancreas, prostate, kidney, and lung[17]and is essential for both embryonic liver development and liver regeneration after injury[18-20].
RECEPTOR TKIS DE-REPRESS INNATE IMMUNE RESPONSES IN TUMOR CELLS AND NORMAL EPITHELIAL CELLS
As previously mentioned, acneiform rash is an established side effect of both small molecule and antibody-based inhibitors of EGFR[21], typically presents within the first two weeks of administration of EGFR inhibitor and is a positive predictor of response to therapy. Not only is there a positive correlation between rash and therapeutic response of the tumor, but progression free survival and overall survival are also positively correlated with presence of these skin toxicities[22-25]. There are three main contributors to EGFR inhibitor induced skin toxicity;damage to the epithelial barrier, loss of antimicrobial mechanisms, and extensive release of in fl ammatory chemokines and cytokines. For the purpose of this article, we will focus on TKI mediated chemokine and cytokine release. The mechanisms contributing to damage to the integrity of the epithelial barrier and antimicrobial defense loss has been previously reviewed[26]. The EGFR inhibitor-induced inflammation is characterized by robust release of chemokines and cytokines that recruit and activate distinct immune cell populations including dendritic cells, macrophages, granulocytes, mast cells and T cells[12,13,27]. Mechanistically, Pastore and colleagues have demonstrated that this in fl ammatory phenotype is largely driven by a type I interferon (IFN) response that occurs in keratinocytes following inhibition of EGFR signaling. A type I IFN response has classically been viewed as an inducer of cell- intrinsic antimicrobial signals that repress the spread of infectious agents,particularly viral pathogens, to neighboring cells[28]. Type I IFNs also function to modulate innate immune responses that occur upon infection to engage the adaptive immune system through increased expression of the chemokines, CXCL10, CXCL9, CCL2, and CCL5. Studies performed by Mascia et al.[13]using primary human keratinocytes treated in vitro with erlotinib demonstrate an increase in secretion of CCL27, CXCL14, CCL2 and CCL5. Furthermore, immunohistochemistry (IHC) staining performed on cutaneous lesions from patients treated with erlotinib show an increase in markers for macrophages, natural killer (NK) cells, dendritic cells,and both CD4 and CD8 positive T cells. Similar in fl ammatory responses are seen in response to the EGFR TKI,gefitinib and the monoclonal antibody cetuximab. Furthermore, in vivo studies where EGFR has been genetically ablated in murine epidermal cells demonstrates an increase in transcripts for CCL2, CCL5 and CCL22 in the skin and circulation within their first week of life, prior to presence of immune cell populations[12,13].

Figure 1. Induction of an inflammatory phenotype in response to RTK inhibition in normal epithelia and carcinomas. RTK: receptor tyrosine kinase; NK: natural killer
The precise cellular and molecular mechanisms underlying the TKI mediated induction of an in fl ammatory phenotype and patient response to therapy remains ill-defined, but we propose that this response originates within oncogenic tumor cells and alters the immune landscape of the tumor microenvironment (TME).Our recent studies indicate that the TKI mediated in fl ammatory phenotype is not restricted to normal epithelial cells, but also occurs in lung carcinoma cells in response to TKI treatment[34]. We observe that oncogenic EGFR and ALK cause a suppression of an IFN like in fl ammatory response, as evidenced by marked induction of both pro-tumorigenic (IL6, TGFB2) and anti-tumorigenic (CXCL10) chemokines and cytokines following treatment of lung cancer cell lines with oncogene specific TKIs [Figure 1]. We also detect induction of these genes in on-treatment patient lung tumor samples. This IFN-like in fl ammatory response is not unique to EGFR inhibitors as it also occurs in response to crizotinib in EML4-ALK positive lung cancer cell line models. Thus, the literature and our preliminary data support a hypothesis that RTKs actively suppress in fl ammatory pathways, both in settings of normal tissue homeostasis and in cancer cells. Moreover, oncogene targeted agents may lead to recruitment of innate and adaptive immune cells for participation in the tumor response.
Treatment with oncogene specific TKIs induces tumor/epithelial cell-autonomous expression of MHC class I and II involved in antigen presentation, CXCL10 involved in effector immune cell recruitment and IL6,TGFB2 and CCL28 which recruit and activate immune suppressive cell types. This in fl ammatory phenotype represents a normal physiological response in normal epithelial cells and is retained in carcinoma cells providing a link between normal epithelial cell homeostasis and tumor therapeutic response. These proteins and factors are postulated to instruct both pro- and/or anti- tumorigenic immune cells and contribute to the degree of therapeutic response observed in patients with lung cancer driven by oncogenic RTKs.
In addition to engaging in paracrine communication with the TME through activation of an IFN like response,EGFR inhibitors also have the potential to in fl uence immune responses by modulating MHC expression and antigen presentation. In the context of cancer immunology, MHC molecules govern interactions between tumor cells and CD4 and CD8 positive T cells by functioning as antigen presenting machinery for tumor specific antigens. Pollack et al.[29], Kersh et al.[30]and Pollack[31]demonstrated that treatment with multiple EGFR TKIs and cetuximab enhanced the induction of MHCI and MHCII seen when primary keratinocytes and malignant keratinocyte A431 cells were treated with IFNγ. Skin biopsies from patients treated with and EGFR inhibitor also demonstrated an increase in epidermal MHCI expression. This response was also observed with erlotinib, cetuximab, and the pan-ErbB inhibitor, dacomitinib, in head and neck cancer cell lines[32,33]. Interestingly, this EGFR inhibitor-mediated induction of MHCI was observed in the absence of IFNγ. Thesefindings support a role for EGFR not only in immune surveillance via immune cell recruitment,but also in immunoediting through increased antigen presentation.
ROLE OF THE TUMOR IMMUNE MICROENVIRONMENT IN DICTATING IMMUNOTHERAPY RESPONSE
inflammation characterized by expression of genes that drive immune cell infiltration has recently come to light as being important in response to immune-oncology (IO) drugs that inhibit the PD1-PDL1 immune checkpoint. Clinical benefit has been observed in carcinomas of the lung, head and neck, and skin, however patients who are never smokers (i.e., ALK, ROS, and RET positive lung tumors) or whose tumors express mutant EGFR, whether PD-L1 positive or negative, have not experienced benefit[35]. In ALK and EGFR mutant lung cancer patients whose tumors tested high for PD-L1, overall response rate following durvalumab treatment was only 0%-14%. These data suggest that some patients within these cancer subgroups may exhibit innate resistance to immunotherapy agents, despite the presence of PD-L1 positivity. To this end, Gajewski and colleagues have proposed that T cell inflammation within the TME serves as a superior predictive marker of sensitivity to immunotherapy, and that tumors with scant T cell inflammation exhibit poor responses consistent with innate resistance[36-38]. In this context, T cell inflammation is associated with activation of IFN response pathways. As support of this, Ayers et al.[39]and colleagues report an IFNγ signature that predicts response to anti-PD1 better than PD-L1 positivity, alone, across multiple cancers.
Despite some evidence for modestly increased response rates in early trials with combinations of TKIs and IO agents in lung cancer patients, there are tolerability and safety challenges arising as a result of severe toxicities[40]. Based on the results of recent trials, combining these two treatment modalities is predicted to yield enhanced frequency and grade of on-target side effects. In support of this, the TATTON trial [Table 1], a multi-arm phase Ib trial investigating osimertinib in combination with durvalumab in patients with EGFR mutant NSCLC, reported an increase in ILD with the combination compared to either drug alone[41]. Likewise, the phase I CheckMate012 trial with erlotinib in combination with nivolumab in EGFR mutant patients reports incidences of discontinued treatment due to pneumonitis as well as hepatic toxicities[42,43]. Furthermore, the CheckMate370 trial, a single arm study to evaluate the safety of nivolumab in combination with crizotinib in patients with ALK positive NSCLC, also reported incidence of severe and fatal hepatic toxicities[44]. Collectively, the early results from these trials indicate that combining TKIs withimmunotherapy exacerbates the frequency and severity of some of the adverse events seen in the clinic,especially the inflammation-driven toxicities, pneumonitis, hepatitis, and pulmonaryfibrosis.
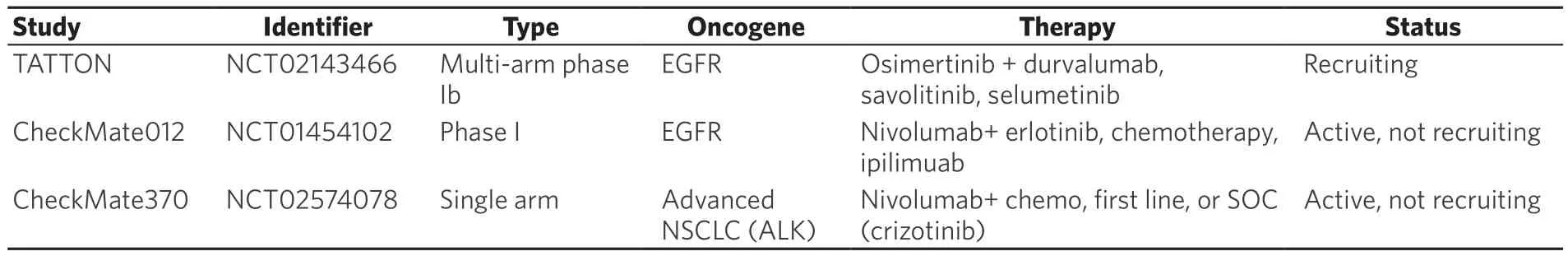
Table 1. Combination therapy trials of TKIs and immunotherapy in lung cancer
The cellular mechanism responsible for less frequent, but more severe TKI-induced adverse events like liver failure and ILD is not defined in the literature. We speculate that it may arise from disruption of normal epithelial tissue homeostasis and an induction of an innate in fl ammatory response similar to that observed with EGFR inhibitors in epidermal tissues. In support, studies demonstrate that genetic disruption of MET in hepatocytes causes an induction of IL6[18]. Livers from hepatocyte-specific MET knockout mice show an increase in immune cell populations including infiltrating neutrophils, macrophages, and cytotoxic T cells[19]. In fl ammatory cytokines also have an established role in hepatocytes, as CXCL10 is found to be expressed by hepatocytes isolated from patients with chronic hepatitis C infection and are correlated with an increase in lobular inflammation and histological severity[45]. Furthermore, the alveolar epithelium is able to contribute to the immune landscape of the lung by generating pro-in fl ammatory cytokines like CXCL10 and CCL2 when stimulated with IFNλ[46]. Studies to investigate the effects of gefitinib on airway repair after injury demonstrated that mice treated with gefitinib after naphthalene induced airway injury developed severe pneumonitis driven primarily by infiltrating neutrophils[47,48]. Bronchial epithelial cells harvested from these mice demonstrated an increase in proin fl ammatory genes. Taken together, these studies suggest that TKIs induce an innate in fl ammatory immune response in epithelial tissues where their RTK targets function as dominant signal pathways controlling epithelial homeostasis. Thus, EGFR or MET blockade may contribute to adverse events like liver toxicities and ILD especially when combined with presently deployed anti-PD1/PD-L1 agents.
CONCLUSIONS AND PERSPECTIVES
Although induction of clinically graded skin toxicities related to an in fl ammatory phenotype has been classified as an adverse event in cancer patients, we propose that this response represents on-target inhibition of a normal tissue homeostasis program in epithelial cells that is retained in their transformed derivatives.Importantly, this TKI-induced innate immune response may actually represent a therapeutic vulnerability for the tumor. The ability of EGFR and ALK inhibitors to stimulate this response in lung cancers driven by mutant EGFR and ALK is clinically relevant considering their poor responses to immunotherapies deployed as monotherapies. We propose that oncogenic RTKs such as EGFR, ALK, ROS1, and MET act to suppress inflammation mediated by this innate immune response and thereby, actively contribute to immune evasion, a hallmark of cancer. Treatment with TKIs counteract this suppression, thereby “releasing the brake” on in fl ammatory signaling pathways and allowing for recruitment of effector immune cells and increased antigen presentation [Figure 1]. This provides a mechanism to explain the connection between an in fl ammatory phenotype and response to TKI. Although this TKI mediated release on in fl ammatory suppression represents a novel vulnerability that may be capitalized on by treatment with IO, early clinical data indicate that combining TKIs with existing IO exacerbates the frequency and degree of some adverse events, especially pneumonitis, hepatitis, and pulmonaryfibrosis. This calls for further preclinical mechanistic studies to fully understand the impact of TKIs on the crosstalk between the TME and cancer cells, as well as the effect on normal epithelial tissue function.
DECLARATIONS
Authors’ contributions
Contributed equally to the preparation of the manuscript andfigures: Both authors Read and approved thefinal manuscript: Both authors
Availability of data and materials
Not applicable.
Financial support and sponsorship
None.
Conflicts of interest
Both authors declared that there are no con fl icts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2018.