Glutamine metabolism in cancer therapy
2018-12-19TraLyNguyenRaDur
Tra-Ly Nguyen, Raúl V. Durán
Institut Européen de Chimie et Biologie, INSERM U1218, Université de Bordeaux, 2 Rue Robert Escarpit, 33607 Pessac,France.
Abstract The amino acid glutamine plays a key role in the metabolism of highly proliferating cells. During malignant transformation, cancer cells modify the consumption and processing of glutamine to sustain cell growth and proliferation.In some cases, these cancer cells become addicted to glutamine. Thus, targeting the metabolism of glutamine has been developed during last years as a potential strategy against cancer. In this review, we summarized the last advances in our knowledge about the role of glutamine metabolism in cancer therapy.
Keywords: Cancer, glutamine, glutamoptosis, metabolism, mammalian target of rapamycin
INTRODUCTION
Since the discovery of the “Warburg effect” about the reprogramming of glucose metabolism in the 1920s,there is a massive interest in understanding cancer metabolism for therapeutic benefits. Glucose and glutamine are two most important nutrients used by cancer cells for their proliferation and growth. While the glycolytic pathway produces ATP and metabolic intermediates for biosynthesis, glutamine metabolism can supply amino acids, nucleic acids and glutathione necessary for cell proliferation. In this review, we describe the metabolic transformation in cancer cells, then summarize different roles of glutamine in cancer metabolism in biosynthesis and stress protection. Finally, we discuss potential cancer therapy targeting approaches based on glutamine metabolism.
METABOLIC TRANSFORMATION IN CANCER CELLS
Among the different hallmarks of cancer[1], metabolic transformation plays a key role in the adaptation of cancer cells to a changing environment. Cancer cells harbor oncogenic mutations, leading to an increase in nutrient uptake, and altering their metabolism to support anabolic processes for cell growth and proliferation.
Uptake
In order to guarantee a rapid cell proliferation, cancer cells first need to increase the uptake of nutrients from the extracellular environment. Glucose and glutamine are two main nutrients that cancer cells uptake from extracellular environment. Cancer cells become easily “addicted” to glucose and glutamine, as their withdrawal can induce cell death. Through the catabolism of glucose and glutamine, the cells produce both carbon intermediates as building blocks and reducing power for macromolecules production and ATP generation. The increase in glucose consumption by cancer cells was first described by Warburg[2]. He saw that cancer cells consume 10 times more glucose than non-proliferating normal cells, and they convert glucose to lactate even in the presence of oxygen and fully functioning mitochondrial respiration. The socalled “Warburg effect” (or aerobic glycolysis) has become a well-known and common metabolic phenotype allowing tumors to fulfil the energetic requirement for cell growth[3]. Positron emission tomography(PET)-based imaging of the high uptake of a radioactive fl uorine-labeled glucose analogue 18F- fl uorodeoxyglucose (18F-FDG) by cancer cells is used as an imaging tool for the detection of several cancers and for the monitoring of treatment response[4]. Cancer cells acquire oncogenic alterations to increase glucose uptake, independently of external stimuli. For instance, PI3K/AKT pathway promotes both the expression of glucose transporter GLUT1 mRNA and the translocation of GLUT1 protein from endomembranes to the cell surface[5,6]. Furthermore, AKT potentiates the activity of hexokinase and phosphofructokinase enzymes, which catalyse rate-limiting steps of glycolysis, in order to induce glucose consumption to branching pathways[7-9]. Additionally, GLUT1 mRNA expression is upregulated by Src or Ras protein, mostly in the presence of two enhancer elements in the gene[10]. Thus, oncogenic signaling pathways, which are often upregulated in cancer, share also another common point to induce glucose import.
High glutamine demand was first described by Eagle[11], when he saw that cultured HeLa cells required 10 to 100 times more of glutamine than any other amino acid. Not only as carbon source, glutamine is also a nitrogen source for de novo biosynthesis of different nitrogen-containing building blocks, such as purine and pyrimidine nucleotides, glucosamine-6-phosphate, and nonessential amino acids. Moreover, glutamine participates in the uptake of essential amino acids from extracellular environment. For example, leucine is imported through the plasma membrane by the amino acid antiporter LAT1/SLC7A5 in coupling with an eラux of glutamine[12]. Indeed, LAT1/SLC7A5 expression has been reported to be increased in several cancer types[13,14]. Due to the high demand of glutamine, this amino acid is also used for imaging based on 18F-labeled glutamine tracers in preclinical and clinical studies, especially when the use of 18F-FDG is not feasible, like in the brain[15,16]. The mechanisms of glutamine uptake regulation are still being identified.The principal regulator of glutamine utilization is the transcription factor c-myc, which is often upregulated in proliferating cells[17,18]. Indeed, c-myc induces the transcription of glutamine transporters, such as SLC1A5/ASCT2, and also promotes the expression of glutamine-catabolized enzymes such as glutaminase 1 (GLS1) and carbamoyl-phosphate synthetase 2 - aspartate transcarbamylase - dihydroorotase (CAD), in order to encourage glutamine uptake by converting glutamine to glutamate[19-21]. In addition, glutamine uptake can be negatively regulated by retinoblastoma (Rb) tumor suppressor family, whose deletion increases glutamine uptake via the E2F-dependent upregulation of SLC1A5/ASCT2 and GLS1[22]. Thus, glutamine consumption is supported by the activity of c-myc and E2F transcription factors which regulate cell cycle,to ensure the cellular access to glutamine for DNA replication.
Metabolic intermediates for biosynthesis
Despite the original idea of Warburg[2]that aerobic glycolysis was originated as a consequence of mitochondrial dysfunction, subsequent studies showed that mitochondria of cancer cells are still functional and able to conduct oxidative phosphorylation. To adapt to a rapid proliferation, cancer cells need building blocks,intermediary metabolites and reducing power as NADPH. Glycolysis can robustly provide these demands,providing glycolytic intermediates which are diverted into branching pathways. A prominent case of a pathway which uses glycolytic intermediates is the pentose phosphate pathway (PPP). Glucose-6-phosphate produced from glucose can be oxidized by glucose-6-phosphate dehydrogenase to generate NADPH and ribose-5-phosphate, necessary for nucleotide synthesis. PPP is often upregulated in tumors and their enzymes are frequently overexpressed in cancer[23,24]. Another important case is the use of glycolytic 3-phosphoglycerate as a precursor for the serine and glycine metabolism through the one-carbon cycle. Several studies have revealed that the gene encoding 3-phosphoglycerate dehydrogenase, the rate-limiting serine biosynthesis enzyme, is amplified in breast cancers and melanomas[25,26]. Serine and glycine metabolism,derived from glycolytic 3-phosphoglycerate, provide advantages for cell growth, such as nucleotide synthesis, DNA methylation, glutathione production and NADPH generation.
After feeding all branching pathways, the excess of glycolytic fl ux is converted to lactate to preserve a sufficient pool of NAD+ for glycolysis and also to avoid the tricarboxylic acid (TCA) cycle inhibition due to excess NADH. Still, a percentage of pyruvate enters the mitochondria, and a great portion of citrate generated at the TCA cycle from this pyruvate will be secreted to the cytosol through the mitochondrial tricarboxylate carrier. Once at the cytosol, citrate is transformed to acetyl-CoA and oxaloacetate, which is converted to malate for mitochondrial anaplerosis[27,28]. Citrate-derived acetyl-CoA is used as a precursor for lipid biosynthesis and protein acetylation.
In addition to glycolytic intermediates, TCA cycle intermediates are also used for biosynthetic precursors accumulation. The first example is citrate-derived acetyl-CoA, whose production is increased by PI3K/AKT-mediated ATP-citrate lyase (ACLY) enzyme[29]. Secondly, the TCA cycle also provides metabolic precursors for the synthesis of nonessential amino acids, such as aspartate and asparagine from oxaloacetate,or proline and arginine from α-ketoglutarate. Then, aspartate is used for nucleotide biosynthesis. Indeed,enabling aspartate synthesis is an essential role of the oxidative phosphorylation in cell proliferation[30,31].
Due to the release of citrate to the cytosol, the maintenance of the pool of TCA cycle intermediates needs additional in fl ux, called anaplerosis. The main anaplerotic source in growing cells is glutamine[32]. In cmyc-transformed cells, glutamine deprivation could disrupt the TCA cycle and induce cell death, which can be rescued by the addition of oxaloacetate or α-ketoglutarate[33]. Glutamine-derived α-ketoglutarate is oxidized into oxaloacetate to maintain the production of citrate. During hypoxia or under certain oncogenic conditions, α-ketoglutarate could be converted directly to citrate (following a reversed TCA cycle), in order to generate the cytosolic acetyl-CoA when glucose-derived acetyl-CoA is insufficient[34].
GLUTAMINE UTILIZATION IN CANCER CELLS
Glutamine is the most abundant free amino acid in the blood, whose circulating concentration is around 0.5 mmol/L[35]. Despite being a nonessential amino acid, glutamine is physiologically an essential source of carbon and nitrogen for cancer cell proliferation. As discussed above, glutamine uptake is increased specifically in cancer cells that have dysregulated oncogenes and tumor suppressors, such as c-myc. Glutamine is catabolized by different enzymes, including GLS, CAD or glutamine fructose-6-phosphate amidotransferase (GFAT). As an anaplerotic source, glutamine is converted to α-ketoglutarate through mitochondrial glutaminolysis. Glutamine is first deamidated to glutamate, in an irreversible reaction catalysed by the enzyme GLS. Then, glutamate is deaminated to α-ketoglutarate by the enzyme GLUD1/glutamate dehydrogenase (GDH) or by several aminotransferases to produce other non-essential amino acids. Subsequently,α-ketoglutarate enters the TCA cycle to replenish the mitochondrial citrate pool. GLS is the rate-limiting enzyme of glutaminolysis, whose regulation is controlled tightly. There are two isoforms of GLS which are encoded by two genes in mammals, the kidney-type GLS1 and the liver-type GLS2. GLS1 is the main isoform expressed in cancer cells and has been shown to be upregulated in a wide variety of cancers, includ-ing breast, lung, cervix and brain[36]. GLS1 is inhibited by its product, glutamate[37]. GDH activity is also increased in tumor cells. Leucine, a key amino acid from a signaling point of view, is an allosteric activator of GDH, inducing the production of α-ketoglutarate and preventing GLS inhibition by glutamate accumulation[38]. As discussed above, glutamine is imported by the transporter SLC1A5, while leucine is taken up through the bidirectional antiporter SLC7A5 which exports glutamine out of the cell. Thus, glutamine modulates glutaminolysis in combination with leucine [Figure 1].
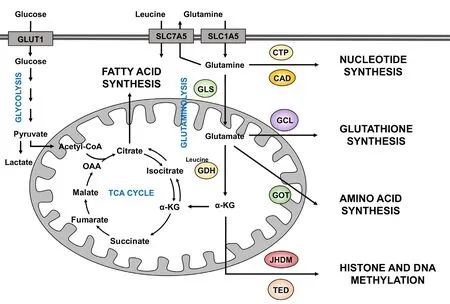
Figure 1. Different uses of glutamine in cancer cells. Glutamine enters the cells through transporters such as SLC1A5. Once inside the cell,glutamine can contribute to nucleotide biosynthesis directly (through CAD for example) or is converted to glutamate by GLS. Moreover,it can also be exported outside of the cell for the import of leucine, a coactivator of GDH. Then, glutamate can be converted to α-KG by GDH. Glutamate can contribute to the synthesis of glutathione through the activity of different enzymes, such as GCL. Amino acid synthesis is supported by the aminotransferases (such as GOT) which converts glutamate to α-KG. Glutamine-derived α-KG can enter the TCA cycle to produce energy for the cell or proceed backwards via the reductive carboxylation to provide an alternative source of lipid synthesis. Moreover, α-KG is a co-substrate of dioxygenase enzymes (such as JHMD and TED) in the regulation of histone and DNA methylation. α-KG: α-ketoglutarate; CAD: carbamoyl-phosphate synthetase 2 aspartate transcarbamylase, and dihydroorotase;CTP: CTP synthetase; GCL: glutamate-cysteine ligase; GLS: glutaminase; GDH: glutamate dehydrogenase; GOT: glutamate-oxaloacetate transaminase; JHMD: Jumonji C histone demethylases; TED: TET DNA demethylases
Glutamine can be synthetized by the cells through GLUL/glutamine synthetase (GS) which catalyses the condensation reaction between glutamate and ammonia in an ATP-dependent manner and generates glutamine. In mammals, GS is mostly expressed in the liver, brain, and muscle. GS has been found to be a marker of hepatocellular carcinoma (HCC) and its elevated expression may enhance the metastatic potential in HCC patients[39]. Moreover, GS expression correlates with a poor survival in glioblastoma patients[40].
Carbon donor
Glutamine-derived carbon incorporation into the TCA cycle is necessary for the bioenergetic needs and biosynthetic precursors of the cells. Glutamine-derived α-ketoglutarate can fuel fatty acids synthesis through the reductive carboxylation mediated by isocitrate dehydrogenase (IDH). IDH catalyzes the oxidative decarboxylation of isocitrate to produce α-ketoglutarate. As this enzyme catalyzes a reversible reaction between isocitrate and α-ketoglutarate, the inverse reaction, so called the reductive carboxylation, could occur to maintain TCA cycle intermediates under mitochondria defects. Emerging evidence reported the role of glutamine mediating reductive carboxylation for lipid biosynthesis and also for redox homeostasis in cancer with dysfunctional mitochondria or under hypoxia[34,41-43].
Nitrogen donor
Glutamine has two atoms of reduced nitrogen, called α-nitrogen and γ-nitrogen. At the level of nucleotide synthesis, glutamine is the nitrogen donor for enzymes in the purine synthesis, including glutamine phosphoribosylpyrophosphate amidotransferase, phosphoribosyl formylglycinamidine synthetase, and guanosine monophosphate synthetase. But glutamine also acts as nitrogen donor, being metabolized by enzymes involved in the synthesis of pyrimidine, including CAD and CTP synthetase. Thus, one glutamine molecule is used in the production of uracil and thymine, two for cytosine and adenine, and three for a guanine base. Besides that, purine and pyrimidine synthesis use also glutamine-derived aspartate,whose supplementation can rescue cell cycle arrest caused by glutamine deprivation[44]. Interestingly, only the γ-nitrogen of glutamine is used for nucleotide synthesis. This nitrogen is also required for the synthesis of NAD, glucosamine-6-phosphate (a precursor for protein glycosylation), and asparagine, a non-essential amino acid that compensates for glutamine deprivation[45].
The α-nitrogen of glutamine is used to produce other non-essential amino acids or polyamines via transamination. This reaction is catalysed by a family of aminotransferases to produce alanine[46], aspartate[47],serine[48], proline[49]and ornithine[50]. Glutamine is the source of at least 50% of non-essential amino acids used in protein synthesis by cancer cells[51]. It is estimated that glutamine represents on average up to 4.7%of all amino acid residues in human proteome, but obviously the percentage can differ from protein to protein[52]. Hence, glutamine is a key structural building block in the biosynthesis of proteins, nucleotides,non-essential amino acids and polyamines to support biomass accumulation and rapid rates of proliferation.
Redox homeostasis control
During tumorigenesis, cancer cells encounter oxidative stress continuously. In order to maintain oxidative homeostasis, the cells need to increase their antioxidant capacity. Glutamine metabolism plays a major role in the cellular anti-oxidative mechanisms. Glutamine-derived glutamate is used in the synthesis of glutathione, through the condensation with cysteine and glycine by glutamate-cysteine ligase and glutathione synthetase. Tracer experiments with labelled 13C-glutamine showed an enrichment of 13C carbons in glutathione. Accordingly, glutamine starvation reduces the glutathione pool of transformed cells[33,53]. Moreover, as cystine is an extracellular source of cysteine, cystine uptake is facilitated by the eラux of glutamate via the xCT antiporter. Once inside the cell, cystine is converted to cysteine, which is then incorporated into glutathione. Indeed, pharmacological inhibition of xCT increases reactive oxygen species (ROS) level and suppresses tumor growth[54,55]. However, different investigations showed that xCT overexpression enhances cell dependency to glutamine or glucose[56-58]. Those studies identified a new function of xCT antiporter as a regulator of nutrient fl exibility by antagonizing glutamine metabolism. Lastly, glutamine oxidation supports redox homeostasis by supplying carbon to malic enzymes, which produce NADPH. Indeed,in proliferating cells, NADPH is used not only for the lipid synthesis, but also for the reduction of oxidized glutathione (GSSG), protecting the cells from oxidative stress[59].
Chromatin organization
Glutamine metabolism does not only generate building blocks and energy for cell growth, but also produces co-substrates for cellular regulatory cascades, including those that regulate chromatin organization.Actually, glutamine-derived α-ketoglutarate is a co-substrate of dioxygenase enzymes, including the TET family and the jumonji (JMJ) family. Enzymes from the TET and JMJ family catalyse histone and DNA de-methylation and they are inhibited by the accumulation succinate, the by-product of these enzymes.
One example of the role of glutamine-derived α-ketoglutarate in the regulation of histone and DNA methylation is the neomorphic mutations in IDH1/2[60,61]. Moreover, loss-of-function mutations of succinate dehydrogenase (SDH) increase cellular succinate level, which inhibits DNA demethylation and contribute to tumorigenesis[62,63]. Finally, low glutamine in the core region of solid tumors led to histone hypermethylation due to decreased α-ketoglutarate level, resulting in cell dedifferentiation and therapeutic resistance in melanoma cells[64]. Accordingly, glutamine metabolism plays a role in gene expression through the contribution of α-ketoglutarate and succinate to chromatin structure modification.
GLUTAMINE ADDICTION IN CANCER
Due to the high demand of cancer cells for glutamine, glutamine metabolism is highly regulated in order to maintain cellular biosynthesis and cell growth. Thus, the machinery which regulates glutamine metabolism, needs to be very efficient to increase the cellular access to glutamine. The first mechanism to enhance glutamine acquisition is to induce glutamine uptake. Different glutamine transporters are known, especially SLC1A5/ASCT2 which is controlled by c-myc or E2F. SLC1A5 is highly expressed in triple-negative breast cancer patients, correlating with poor survival in tumor-bearing mice[65]. Besides, other transporters such as SLC38A1/SNAT1 and SLC38A2/SNAT2 can compensate for the depletion of SLC1A5/ASCT2 to contribute to glutamine uptake[66].
The expression and activity of glutaminolytic enzymes, GLS and GDH, are also tightly regulated. GLS is inhibited by its product glutamate or by inorganic phosphate. Sirtuin 5 (SIRT5), which is overexpressed in lung cancer, decrease the succinylation of GLS to regulate ammonia production and ammonia-induced autophagy[67]. The transcription factor c-myc induces the expression of GLS through the repression of miR-23a and miR-23b62. Furthermore, additional mechanisms are reported to regulate GLS, such as RNA-binding protein regulation of alternative splicing[68,69]or protein degradation through the ubiquitin ligase complex APC/C-Cdh1 during cell cycle progression[70].
Similar to GLS, GDH expression and activity are controlled by different effectors. GDH is allosterically regulated by activators like ADP and leucine, or by inhibitors like ATP, GTP and palmitoyl-CoA[71-73]. At the level of post-translational modification, the sirtuin SIRT4 ADP-ribosylates and downregulates GDH in beta-pancreatic cells, thereby decreasing insulin secretion in response to amino acids during caloriesufficient conditions[74]. When the extracellular glutamine level is limited, some cancer cell lines are able to induce GS expression in order to escape from glutamine deficient-induced cell death. GS has been found to be overexpressed in some cancers, such as breast cancer or glioblastoma, promoting cell proliferation[40,75].GS transcription is activated by different oncogenic pathways, such as PI3K-PKB-FOXO pathway[76], cmyc[77], and Yap1/Hippo pathway[78]. Moreover, GS is inactivated by extracellular glutamine because the presence of glutamine induces GS acetylation by p300/CBP protein, facilitating its ubiquitination and proteasomal degradation[79-81].
Glutamine addiction appears when cancer cells undergo cell death in conditions of glutamine limitation or when glutamine metabolism is inhibited. Many cancer cells which rely on glutamine catabolism for building blocks and energy have been reported to be addicted to glutamine[33,82-84]. Glutamine-addicted cells exhibit a decreased survival, or even undergo apoptotic cell death, associated with an increased in DNA damage, an overproduction of ROS or a decreased reduced/oxidized glutathione (GSH/GSSG) ratio.In this context, the oncogenic transcription factor c-myc plays a key role in the induction of glutamine addiction[18,33]. Together, these results suggested that this phenotype could be exploited as cancer therapy through the use of inhibitors of glutaminolytic enzymes or treatment which induce glutamine depletion like L-asparaginase.
On the contrary, some cell types show glutamine independence due to the expression of GS. Indeed, glioma cells can synthetize glutamine from glutamate through the activity of GS, maintaining the cell proliferation during glutamine deprivation[85]. Also, those cells use glucose as a source for TCA cycle anaplerosis,which can sufficiently provide α-ketoglutarate for glutamate and glutamine synthesis. However, the source of the free ammonia necessary for glutamine synthesis is not clear. Alternatively, some cell types can adapt to glutamine withdrawal using asparagine[45,86]. Asparagine is indeed playing a role in the exchange of extracellular amino acids, especially serine, arginine and histidine[87]. Despite that asparagine is synthetized from glutamine through asparagine synthetase, how cancer cells adapt their metabolic needs during glutamine deprivation remains to be elucidated.
GLUTAMINE METABOLISM AND MTORC1 PATHWAY
Glutamine metabolism and mammalian target of rapamycin complex 1 (mTORC1) pathway have a tight connection through different mechanisms. The activation of mTORC1 by glutamine and other amino acids is mediated by the Rag GTPase pathway. In addition, glutamine plays a role as the eラux solute for the import of leucine which supports glutamine to activate mTORC1 through glutaminolysis. Moreover, glutamine and leucine cooperate to produce α-ketoglutarate through glutaminolysis, which ultimately activates mTORC1. Indeed, short-term glutaminolysis induces mTORC1 lysosomal translocation and activation via the Rag GTPase, then inhibiting autophagy and promoting cell growth[88]. Moreover glutaminolysismediated mTORC1 activation required prolyl hydroxylase (PHD) enzymatic activity in a HIF-independent manner[89]. Those evidence highlight the role of glutaminolysis-PHD-mTORC1 axis in cancer growth. Besides, glutamine stimulates lysosomal translocation and activation of mTORC1 via the small GTPase ARF1 and v-ATPase in RagA and RagB knockout cells without Ragulator contribution[90].
In agreement with this positive connection between glutaminolysis and mTORC1, FOXO-mediated expression of GS inhibits mTOR signaling by blocking its lysosomal translocation[76]. This mechanism is important for maintaining autophagy during nutrient deprivation. Hence, mTORC1 sense glutamine availability in both directions: when glutamine is available, mTORC1 is activated via α-ketoglutarate production; but mTORC1 is inactivated when glutamine production is triggered.
The connection between glutamine metabolism and mTORC1 present additional connection branches, as glutamine also plays a role in autophagy-induced mTORC1 restoration during amino acid starvation[91].Thus, glutamine recycling, supported by autophagy, is sufficient to reactivate mTORC1 under restrictive conditions.
However, and paradoxically, long-term glutaminolysis activation during nutritional restriction induces an unbalanced activation of mTORC1 during nutrient deprivation and promotes apoptosis[92]. This type of metabolic-induced cell death is called “glutamoptosis”, which supports a tumor suppressor role of glutamine metabolism and mTORC1 (normally known as pro-proliferative inducers) during nutritional imbalance. During glutamoptosis, mTORC1-mediated inhibition of autophagy leads to the accumulation of the autophagic cargo protein sequestosome1/p62 (SQSTM1/p62). Then SQSTM1/p62 interacts with Caspase 8 and activates it to trigger apoptosis. Strikingly, the inhibition of mTORC1 by rapamycin promoted cell survival upon amino acid starvation, which could partially explain the resistance to rapamycin treatment observed in some tumor cells.
Conversely, mTORC1 can regulate glutamine metabolism via different mechanisms. GLS and GDH are both regulated by mTORC1 pathway. Mechanistically, mTORC1 inhibits the transcription of SIRT4 by degrading its activator CREB2 (cyclic adenosine monophosphate responsive element-binding 2), thereby activating GDH[74,93,94]. Also, mTORC1 activate GLS through S6K1/eIF4B-dependent mRNA translation of c-myc, leading to GLS expression by repressing miR-23a/b[20,95]. Intriguingly, in an organotypic 3D tissue culture model, mTORC1 supports the expression of aminotransferases and the suppression of GDH in proliferating cells[96]. Thus, the regulation of glutamine metabolism by mTORC1 is cell type-dependent and needs to be elucidated further. Moreover, mTORC1 controls glutamine transporters SLC1A4 and SLC1A5 expression, thereby promoting glutamine uptake upon androgen receptor signaling in prostate cancer[97].Interestingly, evidence has shown that glutamine fl ux through glutamine transporters activates mTOR signaling[98].
In summary, glutamine uptake and metabolism have a tight connection with mTOR signaling. As both pathways are upregulated in many cancers, strategies which target both glutamine metabolism and mTORC1 signaling have shown synergistic effects against cell growth and proliferation[99].
THERAPEUTIC APPLICATIONS
Given the dependence of cancer cells on glutamine metabolism, targeted therapies have been developed against glutamine metabolism, from glutamine uptake to glutamine-catalysed enzymes. The inhibition of GLS got the attention due to the dysregulation of GLS in a variety of cancers. Indeed, GLS inhibitors have shown promising tumor-suppressive activities in preclinical models for 968 and bis-2-(5-phenylacetamido-1,3,4-thiadiazol-2-yl)ethyl sulfide (BPTES), or even in clinical models for CB-839[100,101]. CB-839 has shown efficacy in triple-negative breast cancer and haematological malignancies therapies[101,102]. In addition to GLS inhibitors, strategies targeting the conversion of glutamate into α-ketoglutarate, such as GDH inhibitors and aminotransferase inhibitors, have also been evaluated in preclinical models of breast cancer and neuroblastoma[103-105].
Nevertheless, most of the compounds are still in the preclinical evaluation stage, or have been directly discarded due to high cytotoxicity. Furthermore, some limitations derived of treatment resistance to targeted therapies against glutamine metabolism have been reported. Induction of pyruvate carboxylase can allow tumor cells to use glucose-derived pyruvate instead of glutamine for anaplerosis, inducing a glutamineindependent growth[106-108]. Also, glutamate-derived glutamine production through GS activity could be another mechanism to overcome glutamine addiction and to promote resistance to glutaminolysis inhibitors[85]. However, combination therapy between glutamine metabolism inhibitors and other pathway inhibitors induced a stronger apoptotic response and enhanced anti-tumor efficacy. For instance, mTOR inhibition in glioblastoma multiforme cell lines led to a compensatory upregulation of glutamine metabolism,promoting mTOR inhibitor resistance. Thus, combined inhibition of mTOR and GLS resulted in synergistic tumor cell death and growth inhibition in xenograft mouse models[99].
CONCLUSION AND FUTURE PERSPECTIVES
Glutamine metabolism plays a central role in the regulation of uncontrolled tumor growth by supplying metabolic intermediates as a carbon and nitrogen source and by maintaining the redox homeostasis against oxidative stress during rapid proliferation. The high demand of cancer cells for glutamine results in glutamine addiction phenotype, which becomes a promising target for the design of new therapeutic strategy. Future investigations will elucidate the molecular mechanism of glutamine addiction by identifying the death pathways activating during the impairment of glutamine catabolism or when glutamine is limited. Finally, the development of an effective drug targeting glutamine metabolism is another challenge for the development of novel anticancer therapeutic strategies.
DECLARATIONS
Authors’ contributions
Wrote the manuscript: Nguyen TL, Durán RV Secured funding: Durán RV
Availability of data and materials
Not applicable.
Financial support and sponsorship
This work was supported by funds from the following institutions: Institut National de la Santé et de la Recherche Médicale - INSERM, Fondation pour la Recherche Médicale, the Conseil Régional d’Aquitaine,Fondation ARC pour la Recherche sur le Cancer, Ligue Contre le Cancer - Gironde, SIRIC-BRIO, Institut Européen de Chimie et Biologie, and University of Bordeaux.
Conflicts of interest
All authors declared that there are no con fl icts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2018.
杂志排行
Cancer Drug Resistance的其它文章
- Sphingolipid metabolism and drug resistance in ovarian cancer
- Molecular bases of Sorcin-dependent resistance to chemotherapeutic agents
- Hitting a moving target: inhibition of the nuclear export receptor XPO1/CRM1 as a therapeutic approach in cancer
- Linking tyrosine kinase inhibitor-mediated inflammation with normal epithelial cell homeostasis and tumor therapeutic responses