超高效液相色谱法测定有色稻米中花色苷的含量
2018-12-15赵珊席清清李曦雷欣宇仲伶俐黄世群罗玲郭灵安周虹
赵珊,席清清,李曦,雷欣宇,仲伶俐,黄世群,罗玲,郭灵安,周虹
(四川省农业科学院分析测试中心,农业部农产品质量安全风险评估试验室(成都),四川 成都,610066)
有色稻米是色素沉积在水稻种子的果皮、种皮或糊粉层内形成的一种重要的特异水稻种质资源,常见的有黑米、红米和紫米。花青素属于黄酮类物质,它是一种广泛存在于自然界植物中的水溶性天然色素,在自然状态下常与各种单糖形成糖苷,成为花色苷[1]。花色苷具有抗氧化活性[2-4]、抗突变活性[5]、抗癌活性[6]、抗炎活性[7-9]、保护视力[10]、抗辐射[11]等功能,在食品、医药、化妆品等行业都具有巨大的应用潜力。有色稻米中含有丰富的花色苷,使得有色稻米的市场需求越来越大。此外,有色稻糠中也富含花色苷,通过了解其花色苷的含量有助于对资源进行更充分的发掘利用。
有色稻米中的花色苷主要有矢车菊素-3-O-葡萄糖苷和芍药素-3-O-葡萄糖苷[12-13]。目前测定花色苷的检测方法主要有可见分光光度法[14]、高效液相色谱法[15]和高效液相色谱-串联质谱法[14, 16]等。高效液相色谱-串联质谱法,其测定准确,检出限低,适合进行低含量样品的测定和鉴定未知结构的花色苷,对于像黑米这类含量较高的样品来说会超过其线性范围。普通高效液相色谱法是目前测定花青素、花色苷较多的方法之一,而超高效液相色谱能够在较宽的线性范围内保持柱效恒定,大幅度改善液相色谱的分析速度、分离度和灵敏度,显著提高工作效率。
本文以黑米、紫米和红米为材料,建立花色苷的超高效液相色谱检测方法。
1 材料与方法
1.1 材料与试剂
黑米稻谷、红米稻谷、紫米稻谷均购于生产基地;矢车菊素-3-O-葡萄糖苷(≥98%)、芍药素-3-O-葡萄糖苷(≥98%),武汉天植生物技术有限公司;浓盐酸(优级纯),西陇化工;乙腈(色谱纯)、甲醇(色谱纯),Fisher Scientific;甲酸(色谱纯),CNW;实验用水均为去离子水。
1.2 仪器与设备
1290型超高效液相色谱仪,美国Agilent公司(配紫外检测器,用于花色苷测定);1100型高效液相色谱仪,美国Agilent公司(配二极管阵列检测器,用于花色苷光谱扫描);KQ5200DE型超声波清洗机,昆山市超声仪器有限公司;AUY220型电子天平,日本岛津公司;UPH-1-20L型实验室超纯水仪,四川优普超纯科技有限公司。
1.3 试验方法
1.3.1 样品制备
将稻谷过砻谷机进行脱壳,分别将脱壳后的糙米和谷壳米糠磨粉机磨成粉并过筛,糙米过80目筛,谷壳米糠过40目筛。-20 ℃避光保存。
有色稻米和米糠花色苷的提取:称取1.0 g(精确到0.000 1 g)糙米粉或米糠样品于50 mL离心管中,加入10 mL提取液V(甲醇)∶V(1.0 mol/L HCl水溶液)=85∶15,混匀后避光超声提取30 min,300 0 r/min离心5 min,取上清液,重复提取1次,合并上清液混匀后过0.22 μm尼龙膜,装入棕色样品瓶中待测。
有色稻米和米糠中花青素的提取:称取1.0~3.0 g(精确到0.000 1g)糙米粉或米糠样品于250 mL具塞三角瓶中,加入50 mL提取液V(无水乙醇)∶V(水)∶V(浓盐酸)=2∶1∶1,混匀后避光超声提取30 min,然后于沸水浴中回流1 h,取出冷却后,用提取液V(无水乙醇)∶V(水)∶V(浓盐酸)=2∶1∶1定容至50 mL后取上清液过0.22 μm水相滤膜,装入棕色样品瓶中待测。具体步骤参照标准NY/T 2640—2014 植物源性食品中花青素的测定高效液相色谱法。
1.3.2 色谱条件
色谱柱:Waters ACQUITY UPLC BEH C18(2.1 mm×100 mm,1.7 μm);柱温:30 ℃;检测波长:520 nm;流动相A:1%甲酸/水溶液(体积比),过0.2 μm水系微孔过滤膜;流动相B:1%甲酸/乙腈溶液(体积比);流速:0.2 mL/min;进样量:2.0 μL;梯度洗脱程序:0~1 min,7%~8% B;1~3 min,8%~18% B;3~4.5 min,18%~30% B;4.5~7 min,30%~40% B;7~8 min,40% B;8~9 min,40%~7% B;后运行2.5 min。
1.3.3 标准溶液的配制
标准储备液的配制:分别称取矢车菊素-3-O-葡萄糖苷、芍药素-3-O-葡萄糖苷标准品各5.0 mg(精确到0.01 mg),用85%甲醇盐酸溶液V(甲醇)∶V(1 mol/L HCl水溶液)=85∶15溶解并定容至10 mL容量瓶中,即为500 μg/mL的单标储备液,于-20 ℃下,贮存于密闭的棕色玻璃瓶中。
混合标准使用液的配制:将单标储备液混合,并用85%甲醇盐酸溶液V(甲醇)∶V(1 mol/L HCl水溶液)=85∶15逐级稀释成0.5、1.0、5.0、10.0、25.0和50.0 μg/mL浓度的花色苷混合标准使用液。
2 结果与讨论
2.1 前处理条件优化
通常花色苷不太稳定,在中性和碱性条件下容易发生分解,但在偏酸性条件下能较稳定地存在。为了解加热酸水解对花青素含量测定的影响,通过比较直接提取测定稻米中花色苷含量和将其沸水浴酸水解后测定花青素含量,结果见表1,发现在沸水浴酸水解过程中花青素有明显的降解。在黑稻和紫稻样品中,矢车菊素测定含量为理论值的57.3%~74.5%,芍药素测定含量为理论值的40.0%~60.4%。因此,选择直接提取稻米中的花色苷后进行矢车菊素-3-O-葡萄糖苷和芍药素-3-O-葡萄糖苷的测定。采用超声提取法对样品中花色苷进行提取(提取溶剂:V(甲醇)∶V(1mol/L HCl水溶液)=85∶15;料液比=1∶10;提取温度:常温),将稻米和米糠样品进行超声避光提取,每次30 min,重复2次。前处理过程迅速、简便。样品处理完后装于棕色的样品瓶中并尽快完成测定。
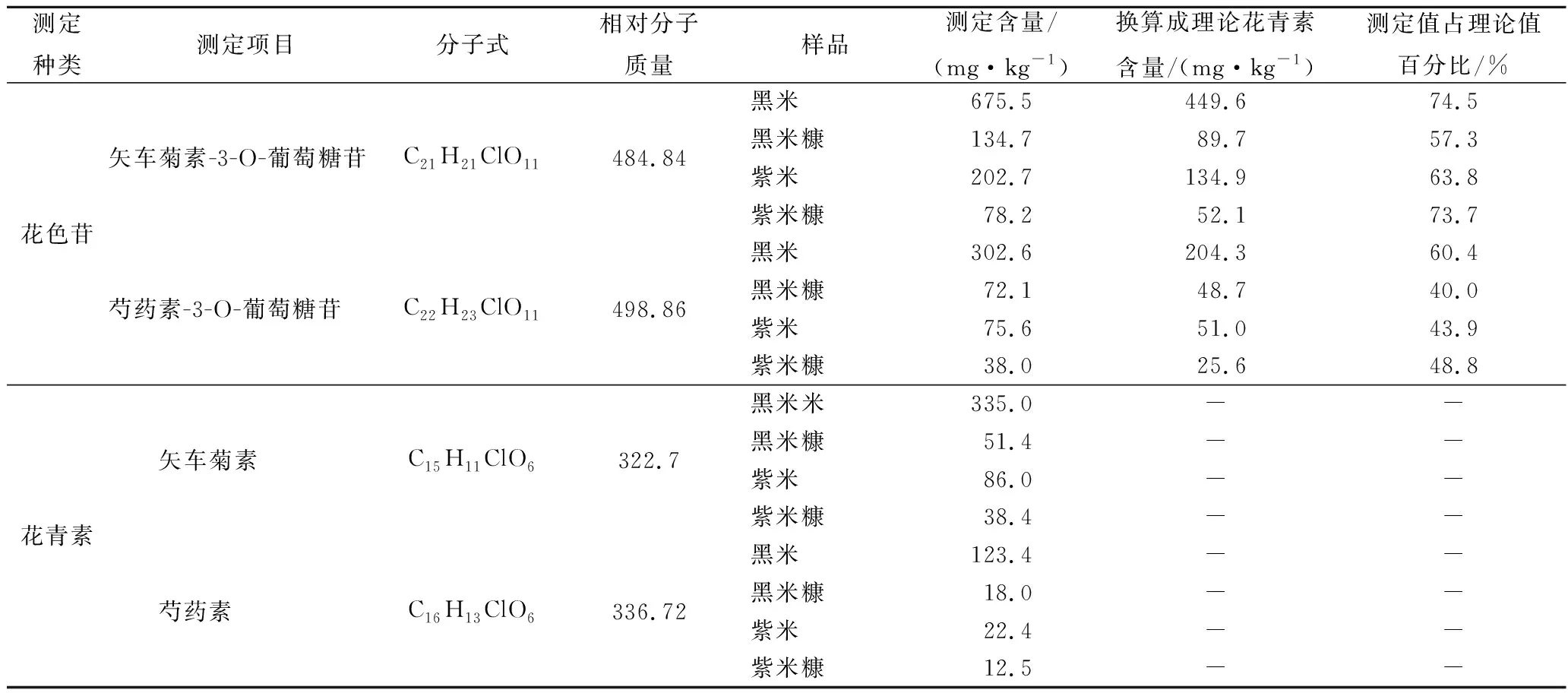
表1 酸水解对花青素含量测定的影响Table1 The influence of acid hydrolysis on anthocyanins content determination
注:“-”表示无数据。
2.2 液相条件的确定
由于花青素需要在低pH值的条件分离,结合文献[17-18]和前期条件摸索,流动相选择酸性较强的1%甲酸水溶液和1%甲酸乙腈溶液。色谱柱选择在低pH条件下稳定性好的Waters ACQUITY UPLC BEH C18色谱柱。考虑到峰形和分离效果,将出峰时间控制在5~7 min,经过反复优化,采用梯度洗脱程序。图1为矢车菊素-3-O-葡萄糖苷和芍药素-3-O-葡萄糖苷的色谱图,A为标准品,B为稻米样品。

A-标准品;B-稻米样品;峰1-矢车菊素-3-O-葡萄糖苷;峰2-芍药素-3-O-葡萄糖苷图1 矢车菊素-3-O-葡萄糖苷和芍药素-3-O-葡萄糖苷色谱图Fig.1 The chromatograms of cyandin-3-O-glucoside chloride and peonidin-3-O-glucoside chloride
2.3 检测波长的确定
本实验采用二极管阵列检测器对花色苷标准品进行最大吸收波长分析,设置扫描范围200~600 nm。提取矢车菊素-3-O-葡萄糖苷和芍药素-3-O-葡萄糖苷的紫外可见吸收光谱图(图2),得到矢车菊素-3-O-葡萄糖苷和芍药素-3-O-葡萄糖苷分别在275 nm和520 nm左右有较强的吸收峰,其中两者在520 nm处吸收强度更强。另外,结合前人的研究结果[16, 19-20],这里选择520 nm为检测波长。
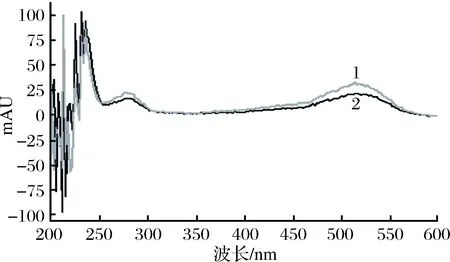
1-矢车菊素-3-O-葡萄糖苷;2-芍药素-3-O-葡萄糖苷图2 矢车菊素-3-O-葡萄糖苷和芍药素-3-O-葡萄糖苷紫外可见吸收光谱图Fig.2 The UV-Vis absorption spectrum of cyandin-3-O-glu-coside chloride and peonidin-3-O-glucoside chloride
2.4 方法的线性、检出限和定量限
将混合标准储备液逐级稀释得到0.5、1.0、5.0、10.0、25.0、50.0 μg/mL浓度的花色苷混合标准使用液,依次进超高效液相色谱仪进行分析,结果表明,在0.5~50.0 μg/mL线性关系良好。仪器的检出限和定量限分别对应仪器3和10倍信噪比,方法检测线和定量限以称样量为1.00 g,定容体积为20 mL进行计算得出。线性关系、相关系数、仪器和方法检出限、定量限见表2。
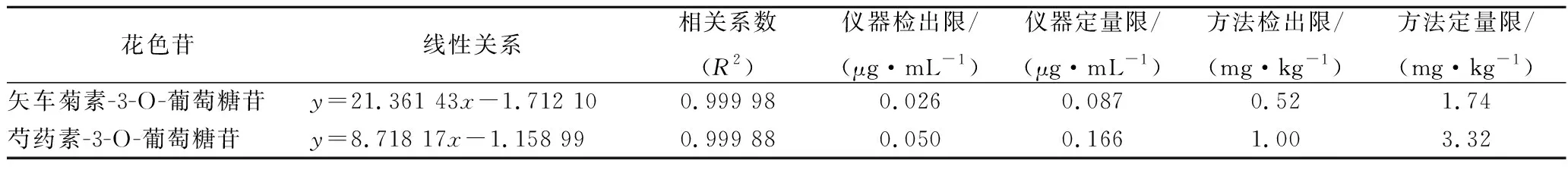
表2 花色苷的线性关系、检出限和定量限Table 2 Liner equations, correlation, LOD and LOQ of anthocyanins
2.5 回收率和精密度
以质量浓度为5.0 μg/mL的混标溶液连续进样6次,根据保留时间和峰面积分别计算精密度,两种花色苷的保留时间RSD分别为0.06%和0.12%,峰面积RSD分别为0.48%和1.27%,表明仪器精密度良好,结果见表3。
为了评价方法的可靠性,选择黑稻米、黑稻糠、紫稻米、紫稻糠、红稻米和红稻糠样品为加标基质进行添加回收实验。结果如表4所示,每个添加水平重复6次。矢车菊素-3-O-葡萄糖苷回收率在93.0%~98.5%,RSD在0.33%~3.50%;芍药素-3-O-葡萄糖苷回收率在96.0%~111.7%,RSD在0.42%~2.18%。结果表明,该方法的准确性与稳定性良好,适用于有色稻中花色苷的检测分析。

表3 精密度实验(n=6)Table 3 Result of precision experiments (n=6)

表4 回收率实验(n=6)Table4 Results of recovery test (n=6)
2.6 样品的测定
本实验对四川重庆地区的4个黑米样品、1个紫米样品和5个红米样品的糙米和米糠进行花色苷的测定,测定结果见表5。由结果可知,黑米中矢车菊素-3-O-葡萄糖苷的含量为0~726.64 mg/kg,芍药素-3-O-葡萄糖苷的含量为0~324.63 mg/kg,总量为0~1 051.27 mg/kg;黑米糠中矢车菊素-3-O-葡萄糖苷的含量为46.90~134.74 mg/kg,芍药素-3- O-葡萄糖苷的含量为15.10~72.08 mg/kg,总量为74.06~206.82 mg/kg。紫米中矢车菊素-3-O-葡萄糖苷的含量为220.00 mg/kg,芍药素-3-O-葡萄糖苷的含量为76.18 mg/kg,总量为296.18 mg/kg;紫米糠中矢车菊素-3-O-葡萄糖苷的含量为78.20 mg/kg,芍药素-3-O-葡萄糖苷的含量为37.95 mg/kg,总量为116.15 mg/kg。红米中均未检出矢车菊素-3-O-葡萄糖苷和芍药素素-3-O-葡萄糖苷,红米糠中矢车菊素-3-O-葡萄糖苷的含量为0~59.94 mg/kg,未检出芍药素-3-O-葡萄糖苷。研究表明[21-22],黑米花色苷分布从米粒表面至内部呈急剧递减,精碾后的精米近乎白米,其花色苷含量极低。陈廷文[23]发现,水稻花色苷含量与谷粒颜色之间有一定的相关关系,颖壳色深褐而糙米色深黑,花色苷含量高。黑米1-米样品未检出矢车菊素-3-O-葡萄糖苷和芍药素素-3-O-葡萄糖苷,可能是由于样品的颜色呈淡黄色,和其他几个黑米样品深黑色差异较大。另外,花色苷类物质在稻米加工过程中,随着米皮去除会有部分损失,因此加工过程越少抗氧化成分保留越多。
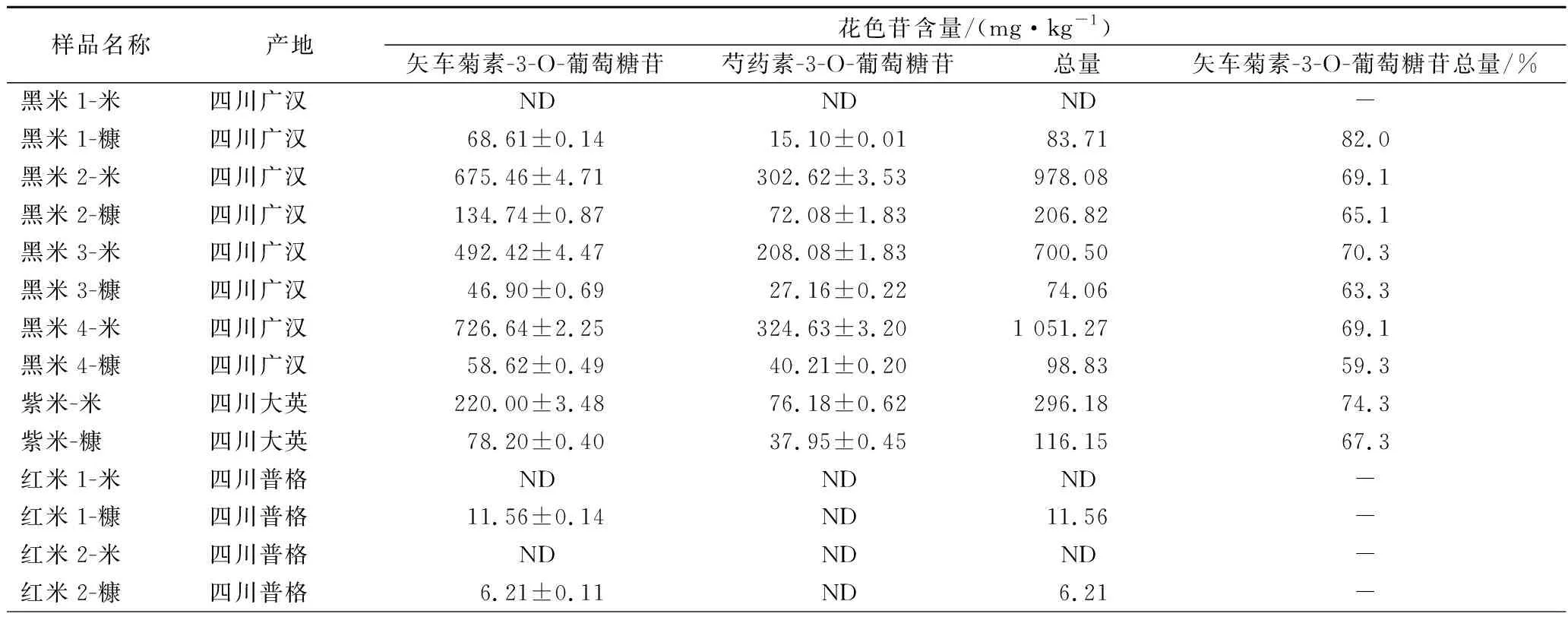
表5 黑米、紫米和红米中花色苷的含量(n=3)Table 5 The content of anthocyanins in black rice, purple rice and red rice
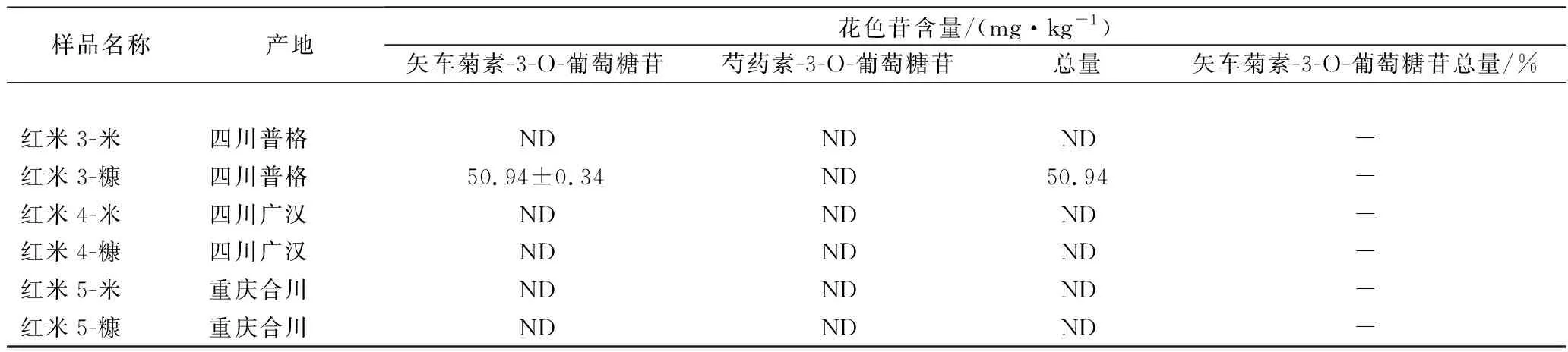
续表5
注:ND为未检出。
不同的黑米种质其花色苷组成和含量都有较大的差异。RYU等[24]从10个不同黑米样品中检测到矢车菊素-3-O-葡萄糖苷和芍药素素-3-O-葡萄糖苷两种花色苷,其总花色苷含量(以全谷计)为0~490 mg/100g。本研究中黑米样品花色苷总量为0~1 051.27 mg/kg,黑米糠样品中花色苷总量为74.06~206.82 mg/kg。本研究发现黑米、黑米糠、紫米和紫米糠中花色苷主要组成是矢车菊素-3-O-葡萄糖苷和芍药素素-3-O-葡萄糖苷,其中矢车菊素-3-O-葡萄糖苷占总量的59.3%~82.0%,这与YAWADIO等[25]和孔令瑶等[26]的研究一致。由不同颜色的水稻样品进行比较得知,黑米的花色苷含量最高,黑米1样品除外,其次是紫米,红米中未检测到花色苷,只有部分红米糠中检测到花色苷。由糙米和米糠样品进行比较得知,大部分黑米和紫米中花色苷的含量大于黑米糠和紫米糠。
3 结论
建立了有色稻米中花色苷的超高效液相色谱检测方法,使用盐酸甲醇溶液超声提取有色稻米花色苷能有效避免花色苷水解等过程发生降解,采用超高效液相色谱—紫外可见检测器对有色稻米和米糠中主要花色苷矢车菊素-3-O-葡萄糖苷和芍药素-3-O-葡萄糖苷进行定量检测。方法验证结果表明,该方法能快速、准确地对有色稻中糙米和米糠的花色苷进行定性定量测定,方法稳定、可靠。
对四川重庆地区的4个黑米、1个紫米和5个红米的糙米和米糠进行花色苷的测定,结果表明:黑米、黑米糠、紫米和紫米糠样品中花色苷主要组成是矢车菊素-3-O-葡萄糖苷和芍药素素-3-O-葡萄糖苷,其中矢车菊素-3-O-葡萄糖苷占总量的59.3%~82.0%。黑米花色苷总量为0~1 051.27 mg/kg,黑米糠花色苷总量为74.06~206.82 mg/kg;紫米花色苷总量为296.18 mg/kg,紫米糠花色苷总量为116.15 mg/kg;红米中未检出矢车菊素-3-O-葡萄糖苷和芍药素-3-O-葡萄糖苷,部分红米糠检出矢车菊素-3-O-葡萄糖苷,含量为0~50.94 mg/kg。
