液相色谱-串联质谱法检测猪肉中20种违禁药物残留的研究
2018-12-15怀文辉倪香艳张晶晶魏紫嫣李文辉孙志文
怀文辉,李 建,倪香艳,张晶晶,魏紫嫣,李文辉,赵 莹,孙志文
(北京市兽药监察所,北京102600)
动物产品中的违禁药物主要包括糖皮质激素、肾上腺激素、性激素及β肾上腺素受体激动剂等,因其具有缩短动物生长周期的作用而常被用于养殖生产中[1]。该类药物具有致突变、致畸形、致癌的潜在危害[2]。研究指出,儿童性早熟、妇女乳腺癌和子宫癌发病率的上升与动物源性食品中性激素摄入有关[3]。 农业部相关公告曾明确规定[2],孕激素、性激素、同化激素及化学合成类激素等在动物性食品中不得检出,在体育比赛中为不得使用的违禁药物。目前,违禁药物残留检测方法主要有酶联免疫法[4]、液相色谱法[5]、气相色谱-质谱法[6]及液相色谱-串联质谱法[7-8]等。酶联免疫法具有较高灵敏度,但容易出现假阳性结果[2];HPLC法抗干扰能力较差,且灵敏度不能满足痕量残留检测的要求;GC-MS法样品衍生化前处理繁琐耗时。LC-MS/MS方法由于无需对样品进行衍生化处理,并具有抗干扰能力强、定性能力强、灵敏度高等优点,是该类药物残留检测的发展方向。但目前尚无同时检测多大类违禁药物残留的高通量检测方法,现有的LC-MS/MS检测方法通常仅针对单类药物的残留检测,检测效率低,成本较高,难以满足不断增加的监管检测任务的要求。
本研究将样品酸水解,以乙酸乙酯提取猪肉中的违禁药物,并以低温冷冻后离心结合固相萃取的方法除脂,建立了猪肉中20种违禁药物残留同时测定的高效液相色谱-串联质谱方法,具有快速、准确的优点,可显著提高违禁药物多残留检测效率。
1 材 料
1.1 仪器 超高效液相色谱-串联质谱仪(美国Waters公司,Xevo TQ-S);电子分析天平(北京赛多利斯有限公司);旋转蒸发仪(上海爱明仪器有限公司);冷冻离心机(美国Sigma公司);氮吹仪(美国Caliper公司);Oasis Prime HLB固相萃取柱(6 mL,200 mg)、固相萃取柱装置(美国 Waters公司)。
1.2 试剂 克伦特罗、莱克多巴胺、沙丁胺醇、特布他林、喷布特罗、西马特罗、甲基睾酮、丙酸睾酮、大力补、群勃龙、睾酮、黄体酮、氟氢可的松、氢化可的松、泼尼松、泼尼松龙、地塞米松、倍他米松、倍氯米松、玉米赤霉烯酮(Dr.Ehrenstorfer公司),纯度≥98.0%。 甲醇、氨水、甲酸、乙腈、乙酸铵、甲酸铵、氢氧化钠、硫酸镁(国药集团)。
2 方法与结果
2.1 溶液的配制
2.1.1 标准储备液的配制 精密称取10 mg克伦特罗、莱克多巴胺、沙丁胺醇、特布他林、喷布特罗、西马特罗、甲基睾酮、丙酸睾酮、大力补、群勃龙、睾酮、黄体酮、氟氢可的松、氢化可的松、泼尼松、泼尼松龙、地塞米松、倍他米松、倍氯米松、玉米赤霉酮、玉米赤霉烯酮、睾酮-D3、黄体酮-D9、玉米赤霉烯酮-D4、玉米赤霉醇-D5、地塞米松-D5、氟氢可的松-D5、克仑特罗-D9、莱克多巴胺-D5、沙丁胺醇-D3,分别置于100 mL棕色容量瓶中,用甲醇溶解并稀释至刻度,配制成浓度为0.1 mg/mL标准贮备液。-20℃避光保存。克伦特罗、莱克多巴胺、沙丁胺醇、特布他林、喷布特罗、西马特罗、甲基睾酮、丙酸睾酮、大力补、群勃龙、睾酮、黄体酮、氟氢可的松、氢化可的松、泼尼松、泼尼松龙、地塞米松、倍他米松、倍氯米松标准储备液有效期6个月。玉米赤霉酮、玉米赤霉烯酮标准贮备液有效期3个月。
2.1.2 标准工作液的配制 精密量取 0.1 mg/mL 标准储备液各1.0 mL,于100 mL棕色容量瓶中,用乙腈稀释至刻度,配制成浓度为1 μg/mL混合标准工作液。 精密量取1 μg/mL混合标准工作液0.5 mL,于10 mL棕色容量瓶中,用乙腈稀释至刻度,配制成浓度为0.05 μg/mL的混合标准工作液。
2.2 样品前处理
2.2.1 样品的提取 准确称取(5±0.05)g 试样于50 mL 离心管中,加入内标工作液 0.1 mL,0.1 mol/L盐酸2 mL,涡旋混匀后50℃超声波处理20 min。以10 mol/L氢氧化钠溶液调节pH 10.0。加入乙酸乙酯20 mL,振荡 10 min。-20℃冷冻 30 min。5000 r/min离心10 min。上清转移至鸡心瓶。下层用乙酸乙酯20 mL如上重复提取1次,离心,合并上清。40℃旋转蒸发至干。加入乙腈氨水溶液(氨水 ∶乙腈 ∶水,3∶5∶92)5 mL溶解残渣,备用。
2.2.2 样品的净化 Oasis Prime HLB固相萃取柱(6 mL,200 mg)依次用甲醇、水各6 mL活化,取全部备用液过柱,用乙腈氨水溶液(氨水∶乙腈 ∶水,3∶5∶92)6 mL淋洗,抽干。以洗脱液(乙酸乙酯 ∶甲醇,6∶4)6 mL洗脱,收集洗脱液,40℃下氮气吹干。加入样品复溶液(乙腈 ∶10 mmol/L甲酸铵溶液,5∶95)0.5 mL溶解残余物,-20℃冷冻30 min。 15000 r/min 离心5 min,上清液过0.22 μm滤膜,供高效液相色谱-串联质谱仪测定。
2.3 仪器测定条件
2.3.1 液相色谱条件 Waters Acquity BEH C18 反相色谱柱(规格 50 mm×2.1 mm,1.7 μm);流动相A:水,流动相 B:乙腈;流速:0.25 mL/min;进样量:10 μL,柱温40℃。梯度洗脱条件见表1。结果表明该色谱条件分离药物效果良好(图1、图2)。

表1 色谱梯度洗脱条件Tab 1 Program of gradient elution
2.3.2 质谱测定条件
2.3.2.1 正离子扫描模式 离子源:电喷雾(ESI)离子源;检测方式:多反应监测;电离电压:2.0 kV;雾化温度:450℃;锥孔气流速:150 L/h;雾化气流速:700 L/h。母离子、子离子质谱参数见表2。结果表明,在该条件下各药物质谱响应灵敏度较高。
2.3.2.2 负离子扫描模式 离子源:电喷雾(ESI)离子源;检测方式:多反应监测;电离电压:2.0 kV;雾化温度:450℃;锥孔气流速:150 L/h;雾化气流速:700 L/h。母离子、子离子质谱参数见表2。结果表明,在该条件下各药物质谱响应灵敏度较高。
2.4 线性关系考察 取空白样品,加入适量内标,经提取和净化后,添加系列标准溶液,制得浓度为1、2、5、10、100 μg/kg 的基质匹配标准溶液,40 ℃氮气吹干。残余物以样品复溶液0.5 mL溶解,-20℃放置30 min。15000 r/min高速离心5 min,取上清过0.22 μm滤膜,供高效液相色谱-串联质谱仪测定。以特征离子质量色谱峰面积与相应内特征离子质量色谱峰面积之比为纵坐标,标准溶液浓度为横坐标,绘制基质匹配标准曲线。结果见表3、表4。 表明本方法在 1~100 μg/kg浓度范围内线性关系良好。
2.5 准确度和精密度考察 准确称取空白样品(5±0.05)g 于 50 mL 离心管,准确加入浓度为0.05 μg/mL 的混合标准工作液适量,配制成添加浓度分别为 0.5、5、10 μg/kg 的样品,充分混匀。 按照2.2~2.4项方法进行测定,每个浓度设5个平行,连续做3 d,计算添加回收率、批内变异系数和批间变异系数,结果见表 5。方法的回收率在63.9%~117%范围内。 批内变异系数 1.9%~9.1%,批间变异系数4.9%~13.1%。表明方法准确度高,精密度较好。
2.6 检出限和定量限的确定 准确称取空白样品,分别准确加入适量混合标准工作液,配制成不同浓度的添加样品。按照2.2~2.4项方法进行测定,根据色谱峰对峰的响应值比值,信噪比(S/N)等于3时的药物浓度确定为方法检出限,信噪比等于10时的药物浓度确定为方法定量限。结果表明,本文建立的检测方法的检出限为0.5 μg/kg,定量限为 1.0 μg/kg。
2.7 样本检测结果 采用本方法对本地屠宰加工企业、养殖厂、大型批发市场猪产品开展样品违禁药物检测。完成40个猪肉样品的检测。氢化可的松和黄体酮有少量检出,浓度较低。
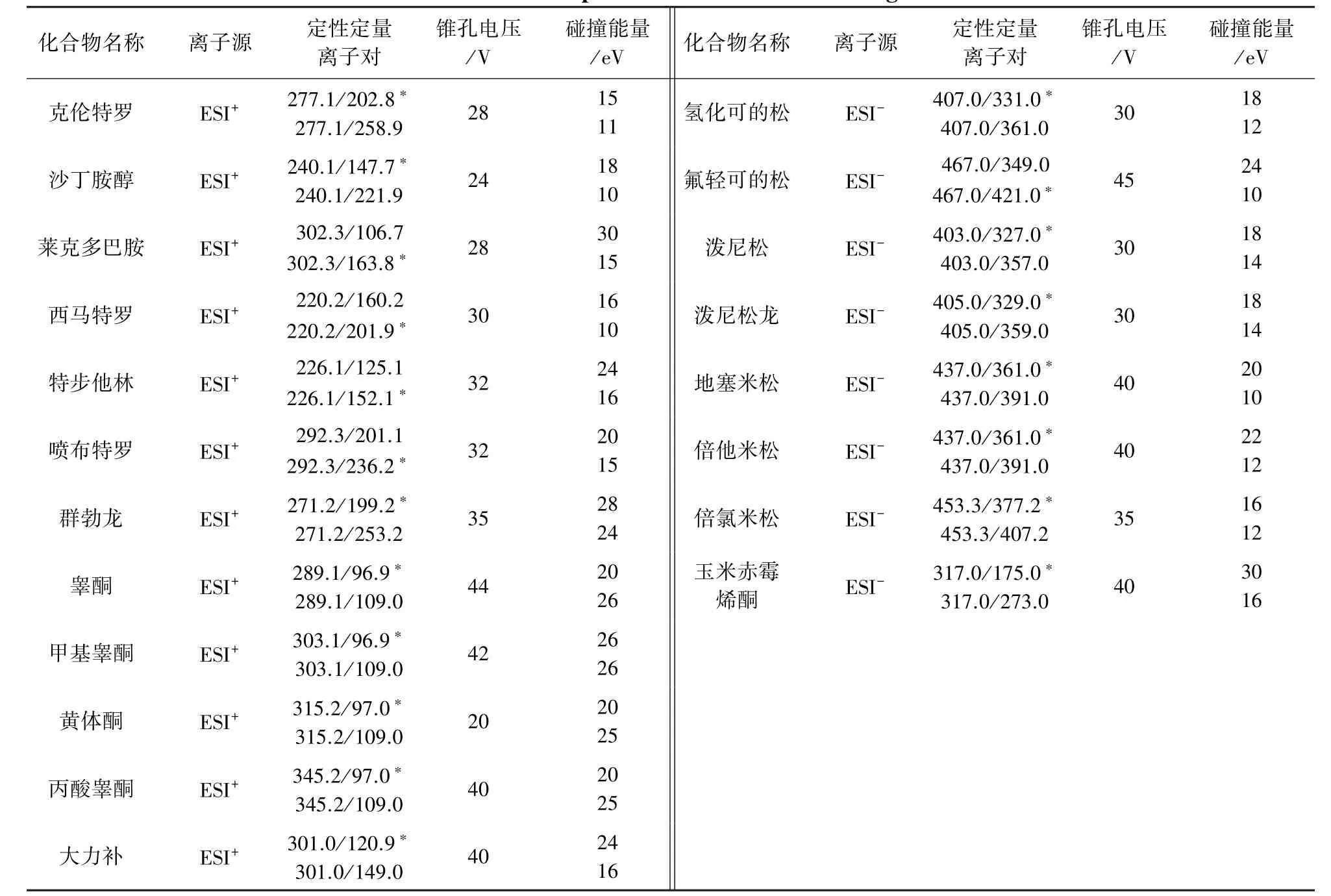
表2 药物MRM参数Tab 2 Mass parameters of 20 banned drugs

表3 药物标准曲线回归方程及相关系数(r2)(ESI+)Tab 3 Regression equations, correlation coefficients of the banned drugs(r2)(ESI+)

表4 药物标准曲线回归方程及相关系数(r2)(ESI-)Tab 4 Regression equations, correlation coefficients of the banned drugs(r2)( ESI-)
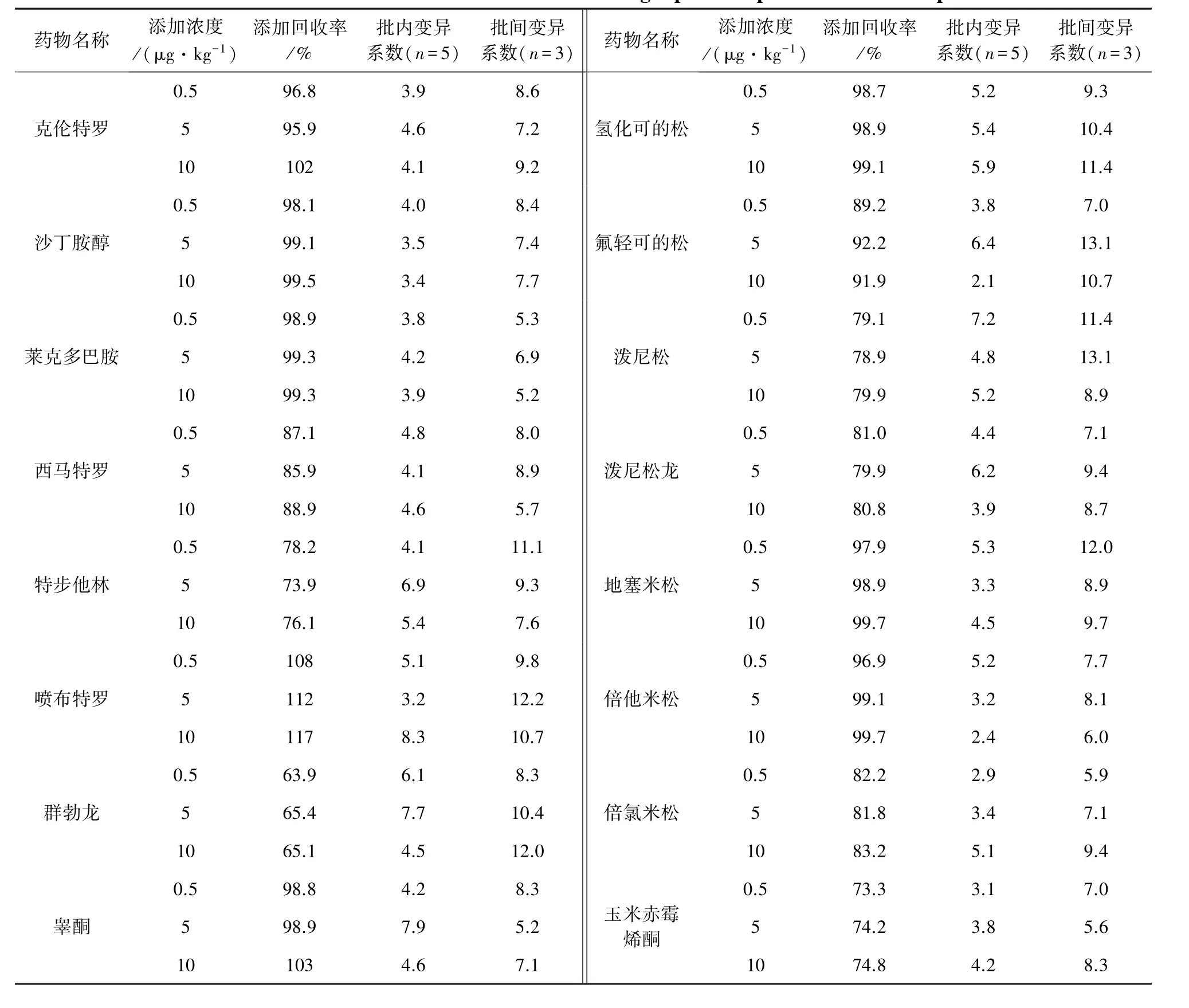
表5 猪肉添加样品检测准确度及精密度Tab 5 Recoveries and RSDs of the banned drugs spiked in porcine muscle samples
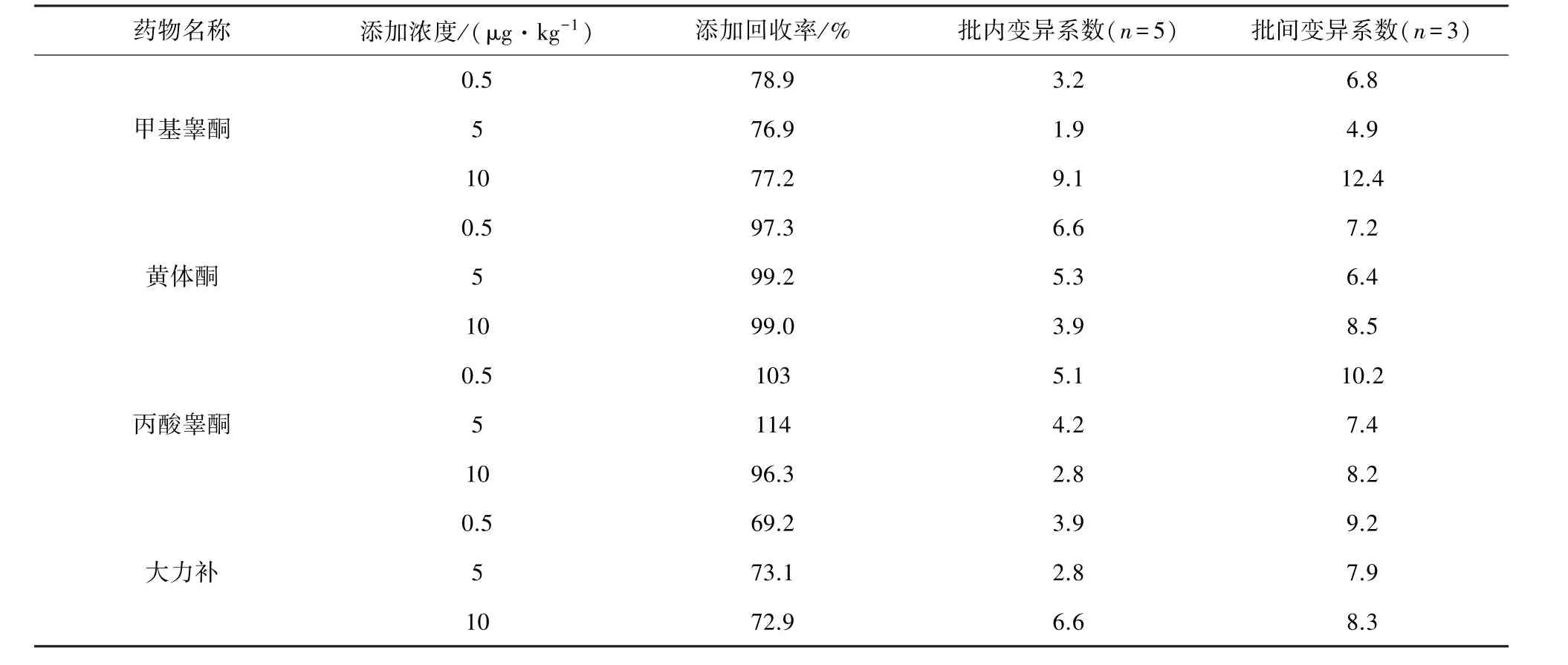
续表

图1 药物混合标准溶液提取离子色谱图(ESI+)(1.0 ng/mL)Fig 1 Extracted ion chromatograms of the illicit drugs(ESI+)(1.0 ng/mL)
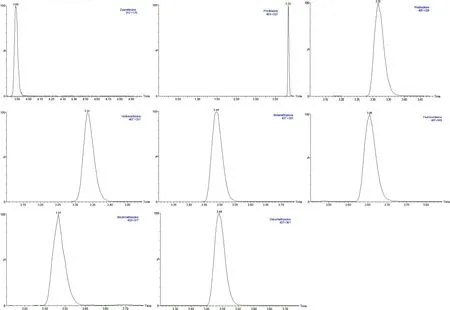
图2 药物混合标准溶液提取离子色谱图(ESI-)(1.0 ng/mL)Fig 2 Extracted ion chromatograms of the illicit drugs(ESI-)(1.0 ng/mL)
3 讨论与分析
3.1 提取方法的选择 违禁药物脂溶性通常较强,可溶于甲醇、乙腈、乙酸乙酯等有机溶剂,易溶于三氯甲烷,但三氯甲烷毒性较大,很少选用。相关研究指出[9-10],乙酸乙酯和乙腈的提取效果较好。考虑到提取过程中杂质干扰和回收率测定,乙腈提取易使样品结团,乙酸乙酯提取则不易结团,振荡混匀效果好,且浓缩过程中耗时短,因此,选用乙酸乙酯为前处理提取试剂。
3.2 净化方法的选择 样品杂质主要为蛋白质、脂类及极性小分子成分。本文建立的方法是同时检测多大类药物残留,提取和检测仪器方法原理存在差异,因此,尽量减低基质效应是方法要解决的关键问题。蛋白质和极性小分子成分可通过有机试剂去除[11]。脂类对检测响应有较大干扰作用,常用去脂方法是正己烷萃取,但药物中同化激素类药物脂溶性强,正己烷除脂会造成部分检测目标药物的损失。根据相关报道[12-13],采用低温冷冻的方法可以有效去除脂类,本文在前处理阶段采用了低温除脂方法。由于部分药物如β-受体激动剂等带有含氮极性基团,有一定的水溶性,为使药物提取充分,在提取液中加入适量氨水以保证得到理想的回收率。为进一步净化,采用了Prime HLB固相萃取柱,该固相萃取柱为新型净化产品,能有效去除磷脂等杂质,并取得较好的净化效果。
3.3 仪器方法的优化 违禁药物的分离多采用C18反相色谱柱[10,14-15]。 本研究采用 C18反相色谱柱分离,以乙腈+水(含10 mmoL/L甲酸铵)作为流动相,梯度洗脱。结果表明以乙腈+水(含10 mmoL/L甲酸铵)为流动相,检测药物有良好的色谱分离效果。同时流动相中加入低浓度挥发性的甲酸铵,可兼顾正离子和负离子模式的质谱响应,色谱峰峰形较优。由于该类药物化学结构有较大差异,导致其对流动相、流速、色谱柱等的响应和灵敏度不同,甚至会出现离子抑制或者增强效应,所以需要考虑兼顾多数化合物的情况,同时进行适当的分段采集分组,以达到满意的效果[10,16]。质谱响应类型分别适于正离子模式和负离子模式下检测。本研究建立的方法,前处理过程通用于两种离子化模式药物的提取,样品液可同时满足正离子模式和负离子模式下的仪器测定。
3.4 样本的测定 采用本文建立方法完成40个猪肉样品的检测。氢化可的松和黄体酮有少量检出,浓度较低,应为内源性来源。根据我国农业部235号公告等法规要求,该类药物不得检出。本研究目的是探索建立猪肉中糖皮质激素类、性激素类、β-受体激动剂类、玉米赤霉醇类药物残留同时检测方法。为全面了解违禁药物的在猪肉中的残留情况,还需要增加采样环节、采样地区以及扩大采样量,进行更多样本的检测。
4 结 论
本研究采用超高效液相色谱-串联质谱技术建立了猪肉样品中多类违禁药物残留的快速筛查方法,对前处理条件、净化方式、色谱和质谱条件等进行了优化,并进行了方法学考察。结果表明,该方法快速、灵敏、准确且稳定;适用于动物性产品中不同离子模式多大类违禁药物残留同时分析检测的要求,可显著提高违禁药物的检测效率。
