误诊为帕金森综合征的肌张力障碍25型1例报告☆
2018-12-11杨改清胥丽霞徐志强张晓艺张东亚
杨改清 胥丽霞 徐志强 张晓艺张东亚
原发性肌张力障碍是一种主动肌与拮抗肌收缩不协调或过度收缩引起的以异常姿势和动作为特征的锥体外系疾病。多数为散发病例,少数有家族史,原发性肌张力障碍的病因可能与遗传、环境等因素有关。目前关于基因突变对该病的致病作用逐渐被发现和证实。肌张力障碍的临床表现主要包括姿势异常、重复动作等,与特发性震颤、舞蹈病、帕金森病等运动障碍性疾病有明显区别,不易误诊。但对于临床表现不典型患者,容易造成误诊和漏诊。本研究报告1例经基因确诊的类似帕金森病表现的肌张力障碍患者,经基因检测出GNAL基因突变,最终确诊为原发性肌张力25型,通过该病例,加深临床医生对肌张力障碍基因诊断的认识。
1 临床资料
1.1 发病情况67岁男性患者,以“运动迟缓半年余,加重2个月”为主诉入院。患者半年余前出现行走缓慢、音调低,2个月余前出现双下肢酸困,抬步困难,站立时身体不自主前倾,间断双足踇趾疼痛,伴颈部僵硬,转身缓慢,曾就诊于加拿大当地医院,给予“骨化三醇胶丸、钙片”等药物治疗,症状曾有好转,但之后逐渐加重,随后回国就诊于多家医院,均诊断为“帕金森综合征”,患者为进一步诊治就诊我院。体型瘦长,睡眠打鼾,间断有睡眠前双下肢抖动。患者于50年前出现双上肢震颤,30余年前出现双下肢震颤。无特殊药物应用及有毒物质接触史。患者父母及姐姐均有双手震颤病史,均在儿童时期起病。
1.2 体格检查查体:体型瘦长,内科查体未见明显异常。神经系统查体:神清,语音低沉,近记忆力下降,反应迟钝,表情减少,四肢腱反射对称亢进,双侧Babinski's征可疑阳性,双踝关节活动笨拙,轮替动作稍僵硬,闭目难立征阳性,间断四肢细小震颤,行动迟缓,前倾体态,双上肢摆臂动作减少,转身缓慢,余神经系统查体未见明显异常。
1.3 辅助检查头颅 MRI(外院):双侧额顶叶、双侧侧脑室旁白质脱髓鞘,双侧基底节区扩大血管间腔,垂体信号欠均匀,双侧上颌窦、双侧筛窦炎,左侧上颌窦囊肿;头颅磁敏感加 权 成 像 (susceptibility weighted imaging,SWI)(外院)示:①右侧基底节区异常信号,考虑钙化或矿物沉积;②右侧小脑蚓部、脑桥、左侧顶叶异常信号,考虑含铁血黄素沉积。头颅磁共振血管造影 (magnetic resonance angiography,MRA)(外院)提示轻度脑动脉硬化改变。入我院后查血常规、血生化、甲状腺功能、甲状腺旁腺激素、血铜蓝蛋白等均正常,中脑黑质超声、脑电图正常,头颅弥散张量成像正常。颈椎MRI:①颈3-4、颈4-5椎间盘突出;②颈椎骨质增生;头颅CT:①右侧基底节区腔隙性脑梗死;②双侧侧脑室旁低密度影,考虑脱髓鞘改变。双足X线:双足第2、3、4中、远节趾间关节间隙狭窄,双足骨质增生。
帕金森病和肌张力障碍基因检测:①未发现受检者SNCA、LRRK2、PARK2、PINK1、PARK7、ATP132A、UCHL1 、GCH1基因存在大片段变异;②患者存在GNAL和POLG基因突变;③患者GNAL基因上的chr18:11752476基因位点上存在c.44G>A杂合突变(氨基酸变化:Gly15Asp,见图1),但患者二姐未发现该基因位点的变异(见图2)。因患者父母已离世,其他兄弟姐妹因在外地定居,未参与基因检测。
1.4 诊断、治疗及随访诊断为肌张力障碍25型,给予“多巴丝肼片62.5 mg每天3次、普拉克索片12.5 mg每天2次”治疗后,1个月后复诊症状较前好转,但患者仍有转身缓慢,调整为“多巴丝肼片62.5 mg每天3次、普拉克索片12.5 mg每天3次”,3个月后、6个月后复诊,患者行动迟缓明显好转。
2 讨论
原发性肌张力障碍是一种不自主、持续的肌肉收缩引起的扭曲、重复动作或姿势异常的综合征,一部分患者可伴有不自主肢体震颤,根据病因分为原发性和继发性,后者多见于外伤、感染、脑血管病等。目前关于原发性肌张力障碍的病因及发病机制尚不清楚。但随着越来越多研究开展,目前对于原发性肌张力障碍的临床表现、环境因素、遗传特点等均有了更深层次的了解,基因因素在该病的发病过程的作用逐渐被研究者注视到、并被很多基础和临床研究逐渐证实[1]。
目前发现的原发性肌张力障碍致病基因已达20种,包括DYT1/TOR1、DYT6/THAP1、DYT4/TUBB4a、DYT7、DTY13、DYT21、DYT23/CIZI、DYT24/ANO3、DYT25/GNAL,大多数为常染色体显性遗传,少部分为常染色体隐性遗传 (DYT5)和 X-连锁 遗传(DYT3),其中C1Z1(DYT23),ANO3(DYT24)和 GNAL(DYT25)是近几年才发现的。 1996年TEZZON[2]第一个发现GNAL基因突变与原发性肌张力障碍有关以来,之后越来越多文献报道了GNAL基因是原发性肌张力障碍25型(DYT25)的致病基因。肌张力障碍患者中GNAL的突变发生率较低,从0到10.52%不等[3],也有报道甚至不到1%[4],发病的平均年龄为31.3岁,且多以颈部肌张力障碍起病。目前已经发现14个GNAL基因突变位点, 包括:c.15C>T、c.30G>T、c.44G>A、c.1059C>T、c.360C >T、c.485T >C、c.142G >C、c.116A >G、c.111C >T、c.111C>T和c.37A>G。突变的方式多样化,包括单核苷酸片段的缺失、错义突变、剪切等[5],这些突变的最终结果一般会导致GNAL所编码的蛋白质结构改变,从而影响到蛋白质的正常生理功能,虽然,GNAL在人群的中的突变率较低,但在很多的种族中均发现这一突变,且多见于成年起病的原发性肌张力障碍患者[1]。
GNAL基因位于18号常染色体的短臂上 (18 p11),GNAL编码的蛋白Gαolf多分布于脑部,尤其是纹状体中[6]。GNAL编码产物为鸟嘌呤核苷酸连接蛋白,又称为Gαolf,由ɑβγ三个亚单位组成,其主要功能是参与多巴胺D1受体的功能和信号传导[7-8]。本病例患者基因突变位点位于chr18:11752476。
原发性肌张力障碍的临床表现主要包括:扭转痉挛、Meige综合征、痉挛性斜颈、手足徐动症、书写痉挛、多巴反应性肌张力障碍、发作性运动障碍等。辅助检查无特异改变。目前尚无公认的肌张力障碍诊断流程,主要依靠病史、特征性的临床表现,并需要结合肌电图、影像学及实验室检查排除其他疾病[9]。目前关于原发性肌张力障碍的治疗,主要包括药物治疗和外科手术治疗,近些年基因治疗被逐渐开始尝试应用到临床病例中,药物治疗主要包括抗胆碱能药物、苯二氮卓类、左旋多巴等。该病例应用含有左旋多巴的多巴丝肼片,反应良好。
对美国2个家庭的基因检测研究[10]显示,GNAL(鸟嘌呤核苷酸结合蛋白G的α亚基)是导致肌张力障碍的致病基因,国内的研究也报道了GNAL基因突变与肌张力障碍25型有关[11]。GNAL基因突变的肌张力障碍患者主要表现为颈部肌张力异常、喉肌张力障碍及头部震颤等,少数全身型肌张力障碍患者也出现了GNAL突变,还有一部分患者会出现颅神经损害的表现或言语障碍,这一现象与国内的研究发现一致[12],因此,一少部分仅表现为震颤的原发性肌张力障碍患者的临床表现与帕金森病较为相识,单纯靠临床表现诊断较为困难。本研究病例主要表现运动迟缓和震颤,与帕金森病、帕金森综合征表现极为类似,结合患者头颅磁共振有多发脱髓鞘改变,因此易被误诊为帕金森综合征,最终通过基因检测证实。本病例GNAL突变的位点位于 chr18:11752476,突变类型为 c.44G>A(图 1),且患者存在颈部僵硬,与肌张力25型临床表型一致。同时,本研究发现患者二姐该位点无杂合突变,且整个GNAL基因序列上均未发现明显突变(图2),患者兄弟姐妹均有肢体震颤,但均未出现活动障碍,因未获得患者父母及其他兄弟姐妹基因检测结果,目前考虑为散发性肌张力障碍,对小剂量“左旋多巴”治疗反应良好。同时患者和二姐均存在POLG基因上 c.3650C>T位点突变(图 3、4),该突变位点为chr15:89860052上缬氨酸替代了丙氨酸。有研究表明,DNA多聚酶 γ(POLG)基因杂合突变是线粒体 DNA缺失综合征4A/4B型、线粒体隐性共济失调综合征、进行性眼外肌麻痹的致病基因[13],但患者及其二姐均无上述三种疾病的临床表现,考虑为无义突变可能性大,但不能除外未来出现上述临床表型可能,需要继续追踪随访患者。

图1 患者GNAL基因

图2 患者二姐GNAL基因
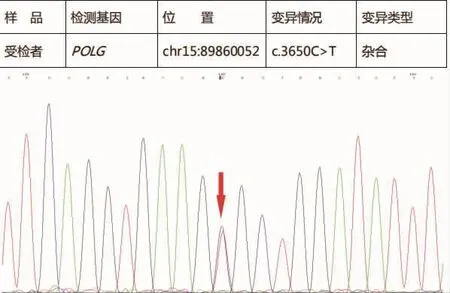
图3 患者POLG基因
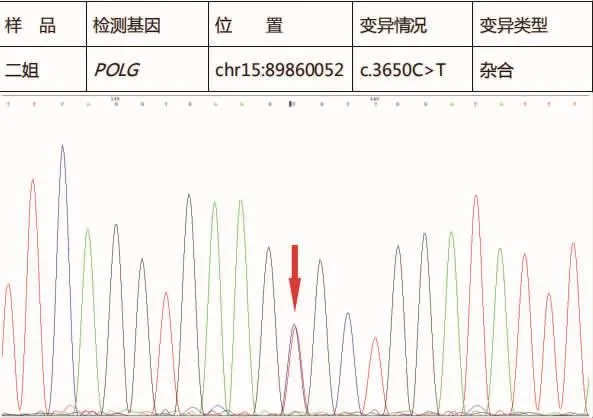
图4 患者二姐POLG基因
某些原发性肌张力障碍在临床表现上类似帕金森病或帕金森综合征,容易导致误诊,因此对于有些临床表现不典型的运动障碍患者,特别是伴有家族性震颤患者,建议行基因检测进行精准化诊治。
