Novel ligand-based docking;molecular dynamic simulations;and absorption,distribution,metabolism,and excretion approach to analyzing potential acetylcholinesterase inhibitors for Alzheimer's disease
2018-12-10SurmniynVijykumrPlniMnogrSrinivsnPrhuRmAvdhrSnjeevkumrSingh
Surmniyn Vijykumr,Plni Mnogr,Srinivsn Prhu,Rm Avdhr Snjeevkumr Singh
aComputational Phytochemistry Laboratory,PG and Research Department of Botany and Microbiology,AVVM Sri Pushpam College(Autonomous)Poondi,Thanjavur(Dist),Tamil Nadu,India
bComputer Aided Drug Design and Molecular Modeling Laboratory,Department of Bioinformatics,Alagappa University,Karaikudi 630004,Tamil Nadu,India
Keywords:Alzheimer's disease Acetylcholinesterase Phytocompounds Molecular docking Free energy calculations Molecular dynamic simulations
A B S T R A C T
1.Introduction
Alzheimer's disease(AD)is an irreversible neurodegenerative disease of brain neurons.The term Alzheimer's was first used by the German physician Alois Alzheimer in 1906[1].According to the World Health Organization(WHO),AD is the commonest cause of dementia,and it affects approximately 25 million people worldwide[2].Acetylcholine(ACh)is an organic substance involved in the transfer of neuronal signals in the brain.Its hydrolysis into a cholineacetyl group is catalyzed by acetylcholinesterase(AChE)[3],which is a well-known enzyme that plays an important role in the central nervous system(CNS)[4].The most important etiological factor in AD is not yet known;however,some established abnormal brain pathologies such as AChE over-expression and extracellular accumulation of“mysterious”β-amyloid plaques are the trademark of this disease.
Currently,four drugs that act as inhibitors of AChE are used in patients with AD,which are tacrine,dopenzial,galantamine,and rivastigmine[5–8].These drugs are known to induce several side effects such as gastrointestinal disturbances,and they have low bioavailability.Hence,it is urgent to develop better inhibitors of AChE from natural sources to treat AD without any side effects.Currently,plants are used to enhance memory and to alleviate other problems associated with AD.Natural products and their derivatives are known to have efficient biological activities against numerous diseases,including CNS disorders[9].Numerous plant-derived substances that may be considered as potential AChE drug molecules belong to different classes of compounds characterized by their structures[10].
Generally,medicinal plant-derived secondary metabolites such as alkaloids, flavonoids,tannins,saponins,and other phytochemicals show promising activities when they are consumed.They are also produced as part of the defence mechanisms against various disorders and some highly pathogenic diseases.These active substances have been found to be beneficial for numerous therapeutic uses[11].Recently,numerous plant-derived drugs have been investigated in clinical phase trials,and the trials of some molecules against globally challenging diseases have been successfully completed[12].
Currently,patients are seeking plant-based drug treatments for their health needs because these drugs are believed to have fewer side effects.Most individuals take only crude extracts for their health complaints which can never let them know the scientific background.Many research studies have isolated bioactive compounds from natural sources,but comprehensive studies are required to determine their molecular interactions.Therefore,in this study,we investigated the efficacy of some bioactive molecules using molecular docking and confirmed their mode of interaction with AChE.
2.Tools and methods
2.1.Modeling platform
The entire computational analysis was carried out using the Schrodinger suite using the Maestro10.2 version packages including LigPrep,Glide XP docking,grid generation,free energy calculations,absorption,distribution,metabolism,and excretion(ADME)toxicity,and MD simulations.Centos Linux was used as the operating system.
2.2.Biological data
In this study,20 bioactive molecules were retrieved from the chemical database[13].The AChE target was taken from the Protein Data Bank[14],and its databank alpha-numeric identity is PDB:1B41.
2.3.Preparation of the protein
The protein was prepared using the wizard tool in Maestro version 10.2.During the process,the missing side and back chains were included[15].The tool has two gears,namely,preparation and refinement.The X-ray crystallography structure of the protein molecules was occasionally bound and tangled with water molecules.The water molecule occupying the protein structure was not suitable for the docking study and,therefore,it was removed.Finally,the optimization and minimization processes were completed in this step[16].
2.4.Ligand preparation
All the ligand molecules were prepared using LigPrep2.4[17],which can generate a number of structures from each input structure with various ionization states,tautomers,stereochemical characteristics,and ring conformations to eliminate molecules on the basis of various criteria such as molecular weight or specified numbers and types of functional groups with correct chiralities for each successfully processed input structure.The OPLS 2005 force field was used for the optimization,which produced the low-energy isomer of the ligand[18].Finally,all the ligand molecules formed in the complex structure for input were docked.
2.5.Molecular docking
First,to test the docking parameters,all the ligand molecules were docked into the binding site of AChE using the Grid glidebased ligand docking program of Schrödinger suite[19,20].In this study,the Maestro10.2 version tool was used to perform rigid, flexible docking for predicting the binding affinity,ligand efficiency,and inhibitory constant to the target[21].The ligands were docked to the active site of AChE using Glide Extra precision(XP),which docks to determine the ligand's flexibility.Only the active small molecule would have available access to avoid the penalties and receive favourable docking scores with accurate hydrophobic contact between the protein and ligand.The electrostatic energy interaction of the hydrogen bonds involved both the side and back chains,hydrophobic contact,and Salt bridge contacts[22].
2.6.MD simulation
MD simulations were carried out using the Desmond software[23,24].The optimized potentials for the liquid simulations(OPLS)-2005 force field were used in this system to determine the protein(AChE)interactions with efficient ligand molecules,which was solvated with the simple point charged(TIP4P)water model[23].The orthorhombic water box was used to create a 10 Å buffer region between the protein atoms and box sides.Overlapping water molecules were deleted,and the systems were neutralized with Na+ions.The OPLS-2005 force field was used for energy calculation.The temperature was maintained constant at 300 K,and a 2.0 fs value was obtained in the integration step.We executed MD simulations for the complex structure of the protein as well as the target with position restraints for 6000 ps to allow the water molecules to remain in the system.Finally,the root mean square deviation(RMSD)was calculated to monitor the stability of the AChE protein in its native motion.The synchronize file was saved every 5000 ps for up to 30 ns and the result was scrutinized using the method described by Nagasundaram et al.[25].
2.7.Prime molecular mechanics‐generalized born surface area(MMGBSA)calculations
The ligand binding energy of each of the 20 phytocompounds required to inhibit AChE was estimated using the prime molecular mechanics‐generalized born surface area(MM-GBSA)module in the Schrodinger Suite 2014[26,27].The total free energy binding(digibind,kcal/mol)was estimated as follows using the software:
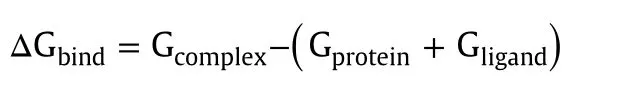
Where each energy term is a combination of G=molecular mechanics energies(MME)+GSGB(SGB solvation model for polar solvation)+GNP (non-polarsolvation)[28]coulomb energy,covalent binding energy,Vander Waals energy,lipophilic energy,GB electrostatic solvation energy,prime energy,hydrogen-bonding energy,hydrophobic contact,and self-contact correction[29].We then used this score to rank the ligand-protein Glide XP docked complex.
2.8.ADME toxicity
This approach has been very useful in drug discovery,especially in determining the mechanism of action of molecules[30].A set of ADME-related properties was calculated for each of the 20 natural compounds using the QikProp program(Schrödinger software),which was run in the normal mode.QikProp generates physically relevant descriptors,and the toxicity of a ligand is considered important for it to act as an effective drug in new drug development[31,32].
3.Results and discussion
3.1.Molecular docking simulations and validation of docking protocol
The current study aimed to exploring the excessive activity of AChE inhibition bioactive molecules.The AChE target had a sequence length of 539 amino acids with a resolution of 2.76 Å.Descriptive hydrogen atoms were added to all the inhibitors to ensure they had all-atom structures,followed by energy minimization.After the preparation process,the protein was ready for molecular docking.This procedure shows the potential of the drug molecules to bind with the protein pocket and their hydrogen bond interactions.The complexes of each of the 20 bioactive molecules docked with the AChE protein.The molecular docking method has produced ligand docking scores with generating their H-bond distance values between ligand and target,and the consequent glide energy was also generated.
In this computational analysis,rutin had a higher docking score than that of the other ligand molecules.Furthermore,it showed a better binding affinity for the target than the other small molecules did.The bioactive molecule of rutin comes under the groups of flavonoids,and it has also exhibited admirable pharmacological and biological activities in numerous experimental studies.Recently,Ganeshpurkar and Saluja[33]reported that rutin shows effective pharmacological potential against the majority of diseases and conditions including neuroinflammation,and it promotes neuralcrest cell survival and has sedative,anticonvulsant,and anti-AD activities.Furthermore,it has been used in treatment of conditions such as hyperkinetic movement disorder,depression,and stroke.
In the present study,most of the bioactive molecules exhibited docking scores above-9.0 Among them,rutin had a superior docking score of-12.335,followed by hesperidin,which had a score of-10.708.At the end of this analysis,the computational toolshowed some important outputs such as the ligand glide energy value and the MM-GBSA calculations(Table 1).Recently,numerous studies have used the molecular docking method because it identifies suitable drug molecules for the target of interest[34–36].In this study,the phytocompounds showed good measurable binding affinities for the target residues.The binding affinities were indicative of the ligand's contribution to and flexibility for the target.The present study also showed the H-bond distances and their contacts types for each molecule.
3.1.1.Rutin
The AChE protein residues interacted with the ligand atoms,and the surfaces were controlled by a complex array of intermolecular interactions.Such interactions depend on both specific interactions at the bindings site and the non-specific forces outside the target binding pocket.Here,the pattern of interaction between AChE and rutin in the complex is shown.We examined the site at which rutin bound to the target and found that it robustly interacted with the AChE residue to form a hydrogen bond.The Asp264,Asn564,His436,Arg327,Pro399,and Glu344 residues were in contact with the ligand atoms.Furthermore,Pro399 formed covalent hydrogen back chain contacts.Asn 264 formed H-bond back chain contacts with the rutin molecule.Arg327,Asn564,His436,and Glu344 formed H-bond side chain contacts with the ligand.His436 formed contacts with the side chain andππstacking contacts with rutin.It had a superior docking score to that of the other bioactive molecules,which is shown in Table 1.The ligand molecule residue contacts,hydrogen bond distances,and the types of contacts are shown in Fig.1.
3.1.2.Hesperidin
Among the 20 ligands,hesperidin showed a docking score that was close to that of rutin at-10.708,and its glide energy values are shown in Table 1.The docked complex was examined,and the residue contacts with the ligand atoms were observed.The interaction plot shows the residue interactions of Thr269,Asp264,Gly265,Arg327,His436,and Pro399.The contacts formed involved different kinds of bonding lines and were back and side chain contacts,as well as hydrophobic contacts with rutin.Complete residues were formed only with the H-bond back chain contacts,except for the His436 residues because it connected the π-π stacking contacts with the ligand.The scrutinized docked complexdetails such as the H-bond distance values,and the types of contacts are shown in Fig.2.Previously,Remya et al.[37]conducted molecular docking studies of AChE using bioactive molecules,which showed that the hesperidin molecule had good binding affinities for AChE.It also had the second highest docking score in this study,similar to the results of this computational analysis.In addition,the anticoagulant activity of hesperidin has been investigated in vitro studies[37,38].
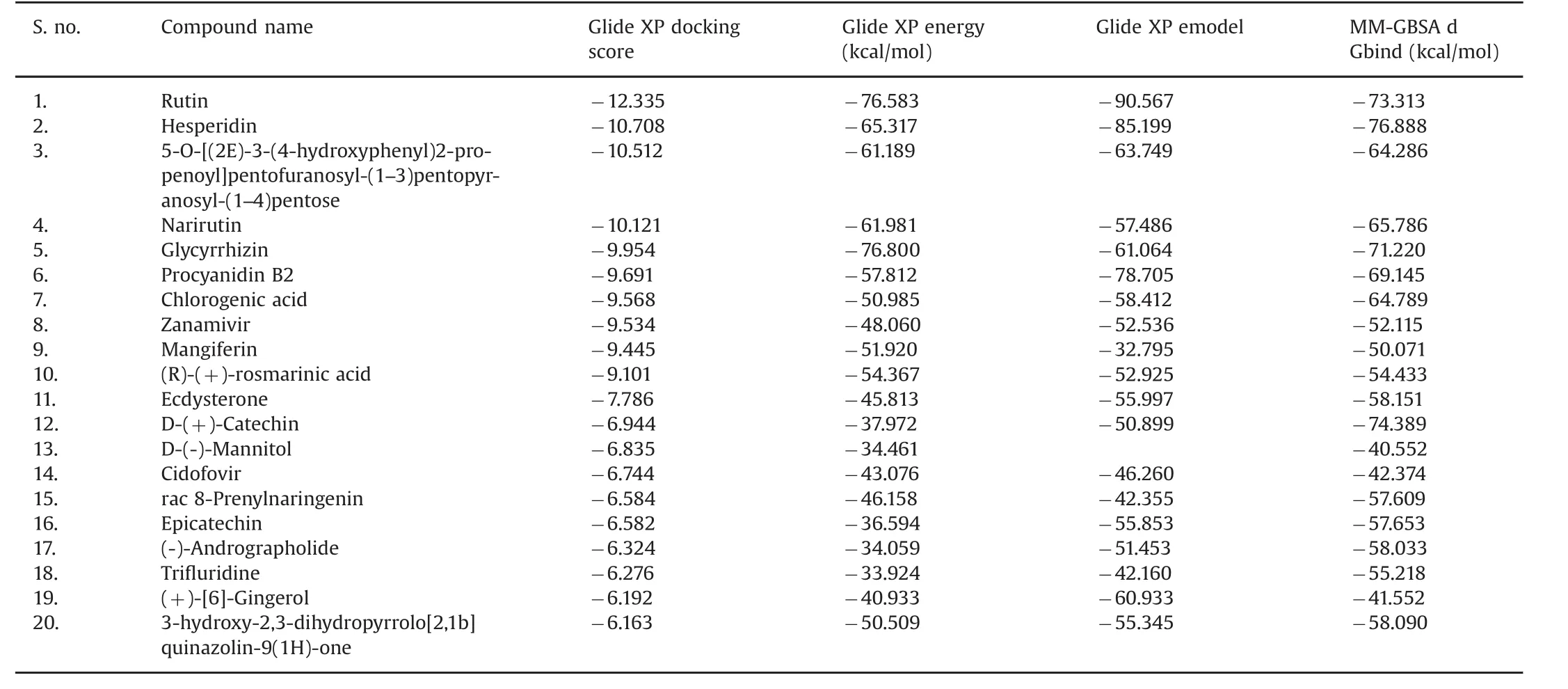
Table 1 Docking results of 20 bioactive molecules(ligands)and the energy generated by the active site of acetylcholinesterase(AChE).
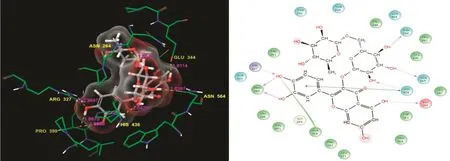
Fig.1.Rutin:Residues and hydrogen bond contacts(yellow dotted line)with their distance values(pink values),and the 2D template representing the types of contacts formed between the ligand and target.
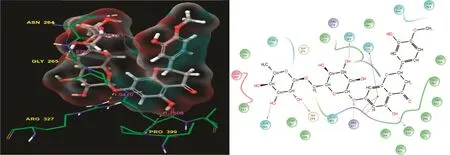
Fig.2.Hesperidin:Residues and hydrogen bond contacts(yellow dotted line)with their distance values(pink values),and the 2D template representing the types of contacts formed between the ligand and target.
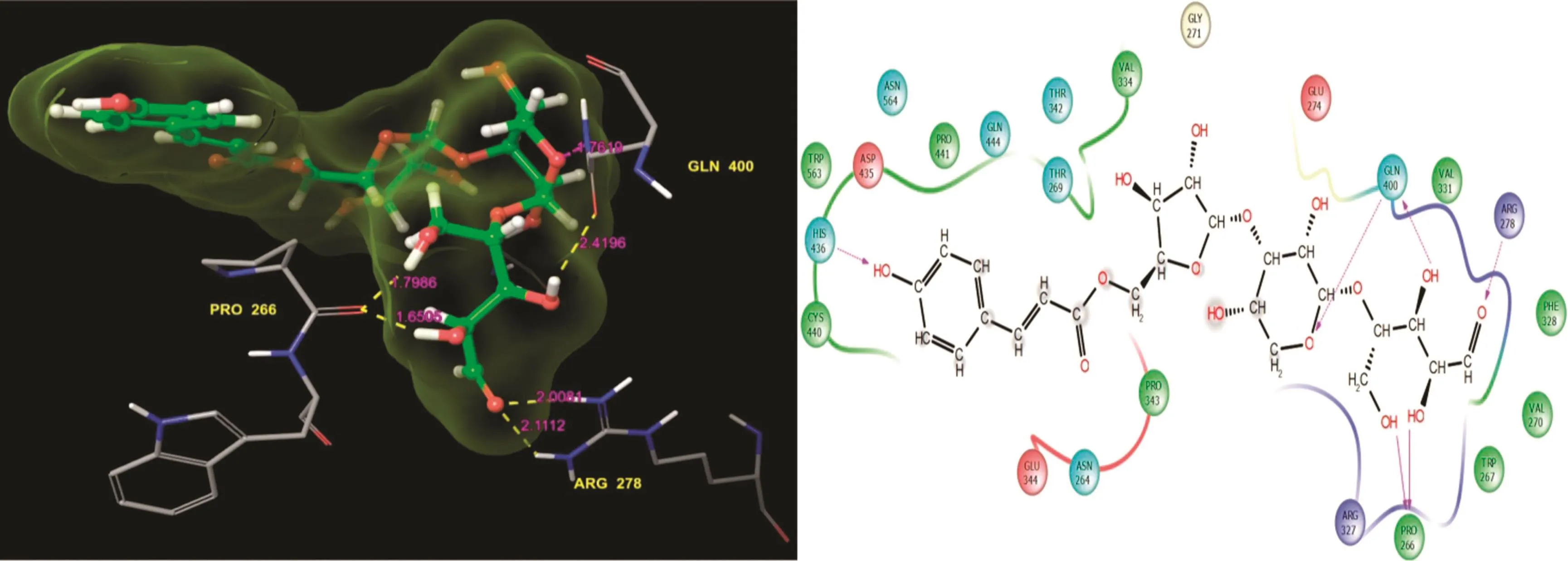
Fig.3.5-O-[(2E)-3-(4-hydroxyphenyl)2-propenoyl]pentofuranosyl-(1-3)pentopyranosyl-(1-4)pentose:Residues and hydrogen bond contacts(yellow dotted line)with their distance values(pink values),and the 2D template representing the types of contacts formed between the ligand and target.
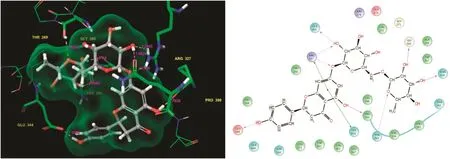
Fig.4.Narirutin:Residues and hydrogen bond contacts(yellow dotted line)with their distance values(pink values),and the 2D template representing the types of contacts formed between the ligand and target.
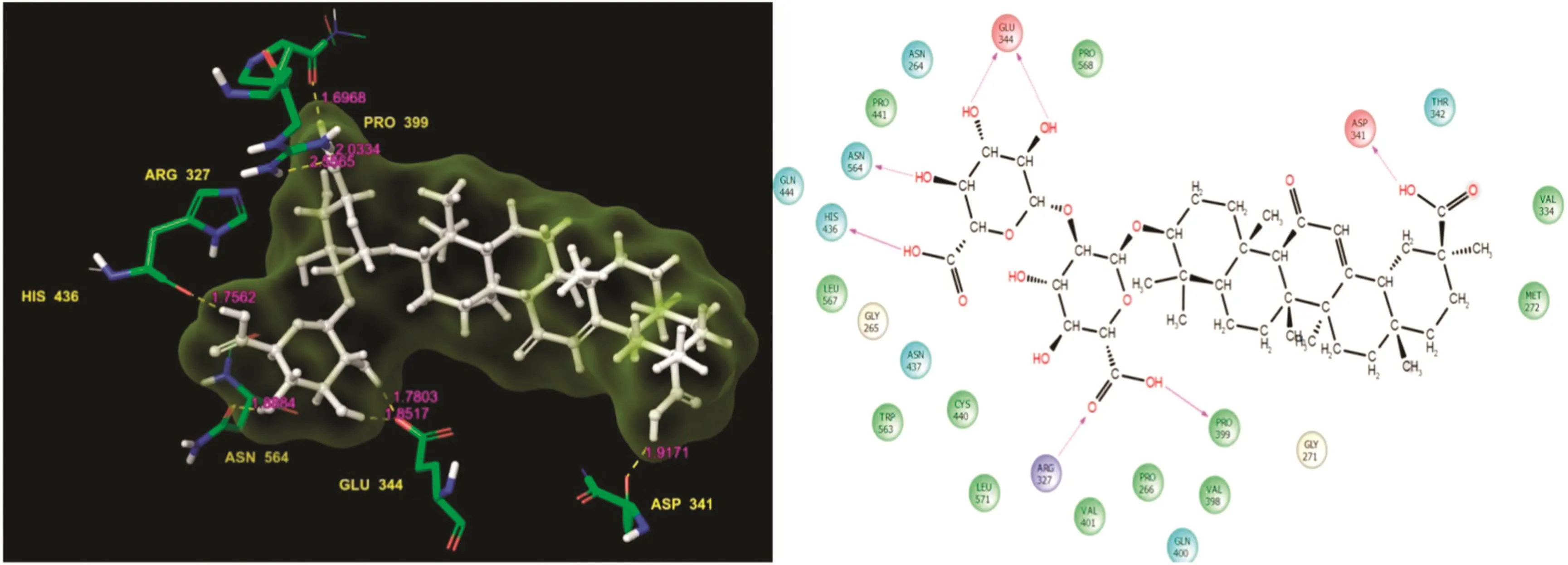
Fig.5.Glycyrrhizin:Residues and hydrogen bond contacts(yellow dotted line)with their distance values(pink values),and the 2D template representing the types of contacts formed between ligand and target.
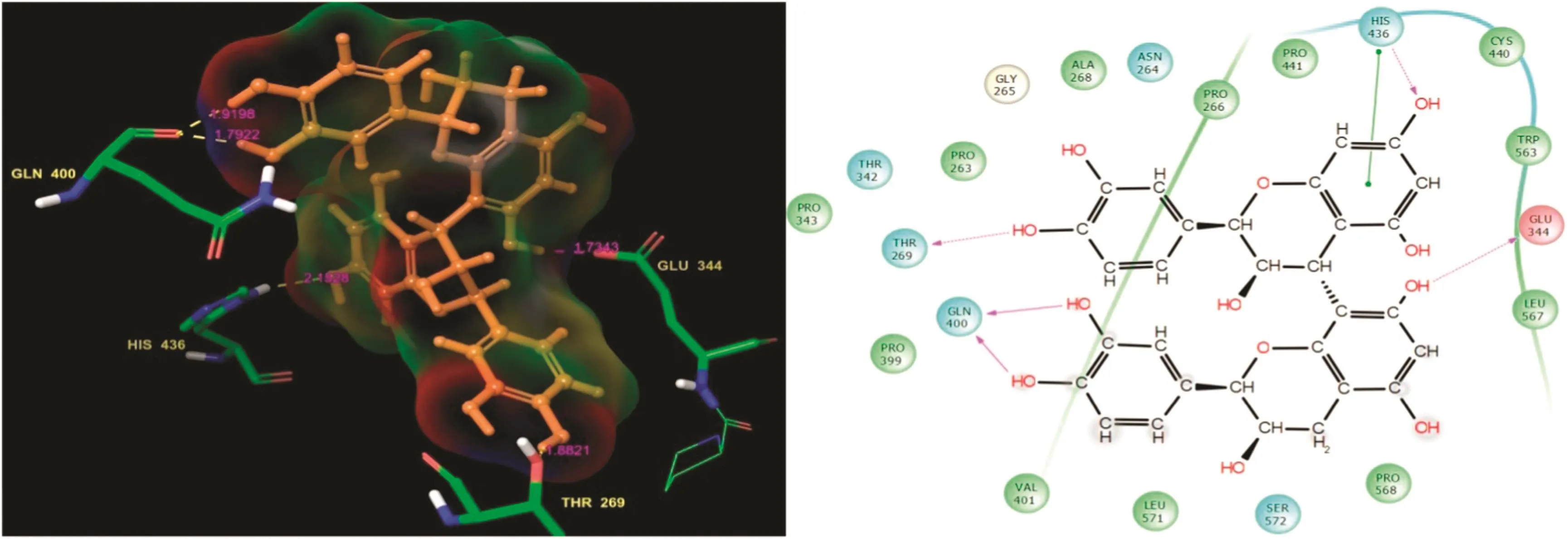
Fig.6.(+)-Procyanidin B2:Residues and hydrogen bond contacts(yellow dotted line)with their distance values(pink values),and the 2D template representing the types of contacts formed between the ligand and target.
3.1.3.5-O-[(2E)-3-(4-hydroxyphenyl)2-propenoyl]pentofuranosyl-(1-3)pentopyranosyl-(1-3.5.4)pentos
The bioactive molecular 5-O-[(2E)-3-(4-hydroxyphenyl)2-propenoyl]pentofuranosyl-(1-3)pentopyranosyl-(1-3.5.4)pentose showed the third most valuable docking score(-10.512)with good glide energy value(Table 1).The docked complex examination showed the residue was in contact with the ligand.His436,Gln400,and Pro266 formed H-bond back and side chain contactswith the ligand.Specifically,the Gln400 residue was covalently bound to the ligand at the side chain contacts,and one end was connected to the ligand oxygen group while the other end was connected to the functional group of the ligand.The remaining Pro266 was covalently bound with the ligand functional group.The ligand molecule residue contacts,hydrogen bond distance values,and the types of contacts are shown in Fig.3.
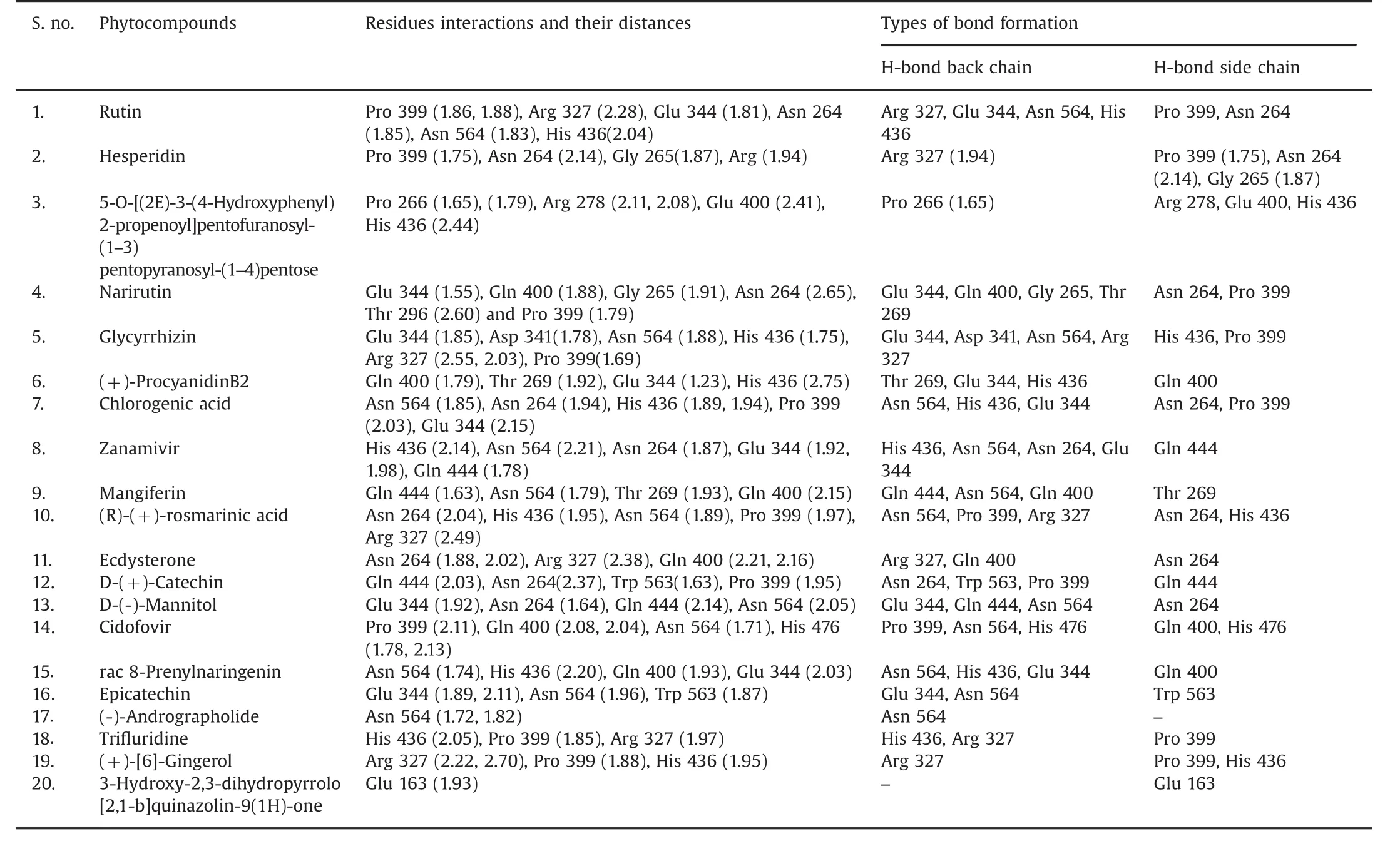
Table 2 Acetylcholinesterase(AChE)enzyme residue contacts with 20 bioactive molecules.
3.1.4.Narirutin
Narirutin had the fourth highest docking score of-10.121 and its glide energy value is shown in Table 1.It showed good binding affinities with the target residues.The scrutinized docked complex clearly showed the residue contacts.Specifically,Glu344,Gln400,Arg327,Thr269,Gly265,and Asn264 formed contacts with various atoms of narirutin.The interaction plot clearly shows the residue contacts with the side chain,back chain,and π-π stacking.Gly265 and Asn264 formed H-bond back chain contacts.The remaining residues,Glu344,Gln400,Arg327,and Thr269,formed H-bond side chain contacts with narirutin.Arg327 formed covalent H-bond contacts with the ligand.One end was connected to the ligand functional group while the other was an oxygen group.The π-π stacking bond contact formation was also shown.The ligand molecule residue contacts,hydrogen bond distance values,and the types of contacts are shown in Fig.4.Murata et al.[39]evaluated the biological potential of narirutin,and found that it showed a significantly strong anti-degranulating activity.
3.1.5.Glycyrrhizin
Glycyrrhizin showed the fifth highest docking score of-9.954 and a good glide energy value,which was the highest in this study(Table 1).It also showed good binding affinity.The docked complex showed a higher level of residue interactions than did the other ligand interaction patterns in this study.Specifically,Asn564,His436,Glu344,Arg327,Pro399,and Asp341 formed different kinds of H-bond contact lines with glycyrrhizin as well as the target.Asn564,Glu344,Arg327,and Asp341 were involved in the H-bond side chain contacts with glycyrrhizin.The Glu344 residue formed covalent contacts with the functional groups of the ligand and its molecule residue contacts,hydrogen bond distance values,and the types of contacts are shown in Fig.5.
3.1.6.(+)-Procyanidin B2
(+)-Procyanidin B2 had the sixth highest docking score of-9.691 and a good glide energy value(Table 1).However,it showed a lower binding affinity than those of the previously listed small molecules.The docked complex interaction template showed it had residue interactions with Thr269,Gln400,His436,and Glu344.Moreover,Thr269,His436,and Gln344 were involved in side chain contacts with(+)-procyanidinB2 while Gln400 formed H-bond back chain contacts.It was also covalently bound with the functional groups of the ligand.Surprisingly,the His436 residue was involved in the H-bond side chain and π-π stacking contacts.The ligand molecule residue contacts,hydrogen bond distance values,and the types of contacts are shown in Fig.6.The AChE residue interactions,H-bond distance values,and their contacts are shown in Table 2.
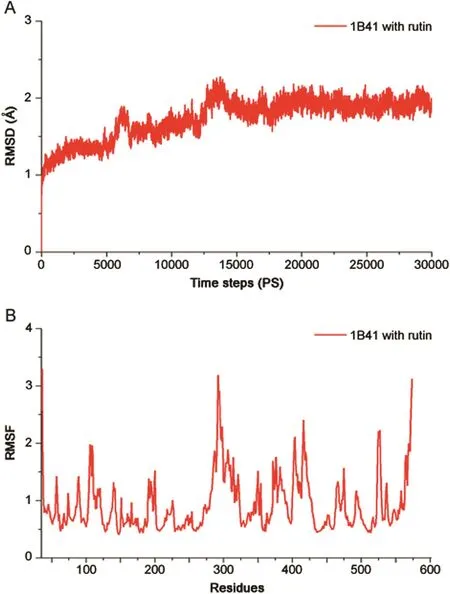
Fig.7.Molecular dynamic(MD)simulations:(A)Root mean standard deviation(RMSD)of rutin with acetylcholinesterase(AChE)complex as a function of simulation time.(B)Root mean square fluctuation(RMSF)values of complex AChE residues with rutin.
Similar to our study,Shruthika and Jency[40]previously performed molecular docking studies.However,they studied bioactive molecules against AD targets.They also screened 13 bioactive molecules to determine their docking scores,binding energy,and number of bonds formed.Based on their research,the bioactive molecules docked against AChE.However,this research outcome represents bioactive molecules that showed superior docking scores and higher maximum binding affinities than those of the molecules reported previously[40].
3.2.MD
The MD simulation was performed for AChE and rutin complex to evaluate the structural constancy using the Desmond software.We ran MD simulations for the AChE protein and rutin for 30 ns.Initially,the RMSD plot showed that the complex deviated for a certain period and attained equilibrium at 17 ns.Subsequently,it remained stable throughout the simulation time for up to 30 ns(Figs.7A and B).
3.3.ADME analysis
In this study,the ADME properties of 20 bioactive molecules were analyzed using the QikProp tool.This analysis presents the physicochemical properties of both synthetic and organic molecules and their biological functions.Based on the previous analysis,the physicochemical and biological properties we analyzed included the bioactive molecules,molecular formula,molecular weight,volume,SASA,acceptor H-bond,donor H-bond groups,the number of ring atoms,QPlogPw(-2 to 6.5),percentage human oral absorption,and CNS effects.These are all listed in Table 3.The ADME-based analysis is an important method for analyzing the efficacy of drug molecules.Much more information is recently available from studies related to ADME toxicity analysis of ligands including donor and acceptor hydrogen bonds,QPlogPw,percentage human oral absorption,and molecular weight[39].
4.Conclusion
AChE is one of the vital enzymes involved in regulating neuronal signaling.The excessive activity of this enzyme in patients with AD causes memory loss and impaired cognitive ability.Patients with AD are currently administered synthetic drugs that affect the organs and induce side effects.Therefore,our research study sought to identify alternative drugs from naturally available plants and their products.Of the bioactive molecules we screened,20 showed potential and were selected.Their activity against the AChE target was analyzed to determine their suitability as drug molecules for treating AD by using computation.Most of the tested ligands exhibited effective dockingscores with good binding affinities.In particular,rutin showed a superior docking score to that of the other bioactive molecules against AChE.Based on the outcome,we further evaluated the residue interaction with rutin and found it showed the maximum level of binding affinities.The other promising bioactive molecules,hesperidin,narirutin,glycyrrhizin and(+)-procyanidin B2 also showed good binding affinities in this analysis.Based on these results,we concluded that rutin is an effective and favourable potential anti-AChE drug and it may be a suitable drug candidate for AD.Further,in vitro studies on the purified rutin molecule are ongoing using various concentrations in neuronal cell lines.
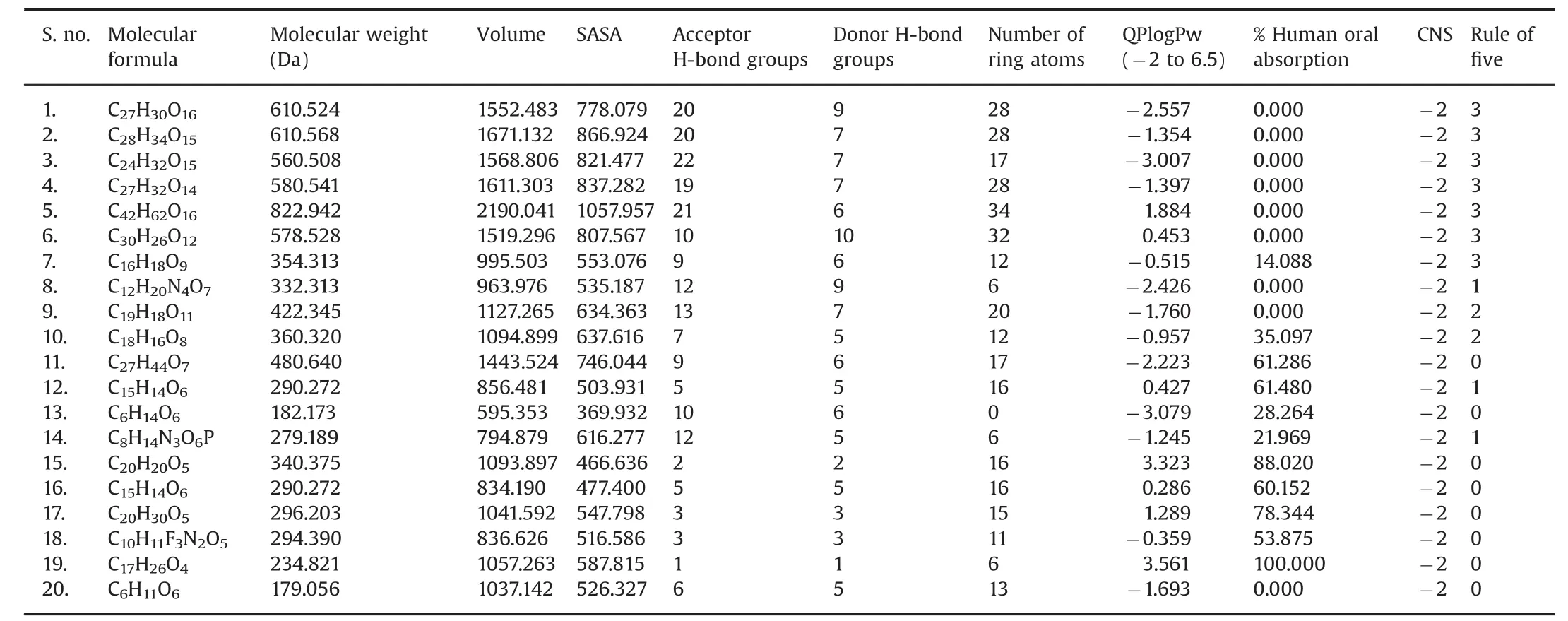
Table 3 Physicochemical properties and biological functions of 20 bioactive molecules analyzed using QuikProp.
Conflicts of interest
The authors declare that there are no conflicts of interest.
Acknowledgments
The authors are grateful to the DST-SERB(SB/YS/LS-109/2014)for providing financial assistance for this project.We especially express our thanks to the management of A.V.V.M.Sri Pushpam College(Autonomous),Poondi,for providing the necessary facilities and support to carry out this work.
杂志排行

Journal of Pharmaceutical Analysis的其它文章
- Recovery rates of combination antibiotic therapy using in vitro microdialysis simulating in vivo conditions
- Evaluation of naproxen-induced oxidative stress,hepatotoxicity and in-vivo genotoxicity in male Wistar rats
- Cytotoxic effect of Rosa canina extract on human colon cancer cells through repression of telomerase expression
- Long-term stability of gentamicin sulfate-ethylenediaminetetraacetic acid disodium salt(EDTA-Na2)solution for catheter locks
- Highly sensitive LC–MS/MS method to estimate doxepin and its metabolite nordoxepin in human plasma for a bioequivalence study
- Detection and determination of undeclared synthetic caffeine in weight loss formulations using HPLC-DAD and UHPLC-MS/MS