Highly sensitive LC–MS/MS method to estimate doxepin and its metabolite nordoxepin in human plasma for a bioequivalence study
2018-12-10NirvPtelMllikSnylNveenShrmDineshPtelPrnvShrivstvBhvinPtel
Nirv P.Ptel,Mllik Snyl,Nveen Shrm,Dinesh S.Ptel,Prnv S.Shrivstv,Bhvin N.Ptel,*
aBio-Analytical Laboratory,Cliantha Research India Ltd.,Bodakdev,Ahmedabad 380054,Gujarat,India
bKadi Sarva Viswavidyalaya,Sector-15,Ghandhinagar 382715,Gujarat,India
cDepartment of Chemistry,St.Xavier's College,Navrangpura,Ahmedabad 380009,Gujarat,India
dDepartment of Chemistry,School of Sciences,Gujarat University,Navrangpura,Ahmedabad 380009,Gujarat,India
Keywords:Doxepin Nordoxepin LC–MS/MS Liquid-liquid extraction Human plasma Bioequivalence study
A B S T R A C T A selective,sensitive and rugged liquid chromatography–tandem mass spectrometry(LC–MS/MS)assay has been developed for the simultaneous determination of doxepin(Dox)and its pharmacologically active metabolite,nordoxepin(NDox)in human plasma.The analytes and their internal standards(IS)were extracted from 500 μL of human plasma by liquid-liquid extraction using methyl tert-butyl ether.Chromatographic separation was achieved on Hypurity C8column(100 mm × 4.6 mm,5 μm)using a mixture of acetonitrile-methanol(95:5,v/v)and 2.0 mM ammonium formate in 93:7(v/v)ratio.Detection was accomplished by tandem mass spectrometry in the positive ionization and multiple reaction monitoring acquisition mode.The protonated precursor to product ion transitions studied for Dox,NDox,and their corresponding ISs,propranolol and desipramine,were m/z 280.1→107.0,266.0→107.0,260.1→116.1 and 267.1→72.1,respectively.A linear dynamic range of 15.0–3900 pg/mL for Dox and 5.00–1300 pg/mL for NDox was established with mean correlation coefficient(r2)of 0.9991 and 0.9993,respectively.The extraction recovery ranged from 86.6%–90.4%and 88.0%–99.1%for Dox and NDox,respectively.The intra-batch and inter-batch precision(%CV)across quality control levels was≤8.3%for both the analytes.Stability evaluated under different storage conditions showed no evidence of degradation and the%change in stability samples compared to nominal concentration ranged from 4.7%to 12.3%.The method was successfully applied to a bioequivalence study of 6 mg doxepin hydrochloride orally disintegrating tablet in 41 healthy Indian subjects under fasting and fed conditions.
1.Introduction
Tricyclic antidepressants(TCAs)are a group of drugs used mainly to treat patients suffering from major depression and other psychiatric disorders including panic disorder,obsessive-compulsive disorder,sleep disorder,eating disorders and attention deficit hyperactivity disorder[1].Doxepin(Dox),a TCA,is widely used for the treatment of depression and has also shown anti-anxiety and anti-histamine properties[2,3].It displays a potent central anticholinergic activity and inhibits the reuptake of noradrenalin and serotonin[4].Among several antidepressant drugs,use of Dox is also associated with suicide and narcotic drug-related deaths[5].Generally,lower dose of Dox has a higher selectivity for H1 receptor and is considered safe for short-term and long-term insomnia[3].Dox is rapidly absorbed from the gastro-intestinal tract and is extensively N-demethylated to its active metabolite nordoxepin(NDox)mainly through cytochrome P450 enzyme 2C19[6].The pharmacological and toxicological properties of NDox are comparable with its parent drug.In addition to NDox,there are several pharmacological lyinactive metabolites that are also formed,including 2-hydroxydoxepin,2-hydroxynordoxepin and doxepine-N-oxide.Dox and NDox are widely distributed throughout the body and their plasma protein binding is about 80%[4].Dox has a plasma life ranging from 8 to 24 h,while the half life of NDox is much longer(>30 h)[7].Therapeutic drug monitoring for Dox is essential due to wide inter-individual variability observed in the pharmacokinetics and in the production of active metabolite.This can help to optimize dose and thus minimize potentially life-threatening toxicity[8].
Bioanalytical methods reported for the analyses of Dox in human whole blood[9],human plasma[4,10]and cerebrospinal fluid[4]include high-performance liquid chromatography(HPLC)[10]and liquid chromatography–tandem mass spectrometry(LC–MS/MS)[4,9].Other methods report simultaneous estimation of Dox and NDox in a variety of biological samples like whole blood[5],gastric fluids[5],bile,urine[5],cerebrospinal fluid[5],tissues[5],hair[7],urine[11]and human plasma[11–13].A majority of these methods were developed for forensic or toxicological studies but only few of them addressed the pharmacokinetics of Dox and NDox in human plasma[11,13].Moreover,they have limited sensitivity (lower limit of quantitation)in the range of 0.25–100 ng/mL for both the analytes.Further,assay methods for the determination of Dox together with several TCAs,other antidepressants and antipsychotic drugs using capillary electrophoresis[14],HPLC[15–19],gas chromatography–mass spectrometry(GC–MS)[20],LC–MS/MS[21,22]and ultra-performance liquid chromatography–tandem mass spectrometry (UPLC–MS/MS)[8,23–25]in biological samples have also been reported.
Based on the work reported thus far,the objective of the present work was to develop and fully validate a selective,sensitive and rugged LC–MS/MS method for the estimation of Dox and NDox in human plasma based on current regulatory guidelines.Further,there are no reports on the pharmacokinetics of Dox and NDox in Indian subjects.The method presents a competent extraction procedure based on liquid-liquid extraction(LLE)to obtain precise and quantitative recovery for both the analytes.The proposed method was successfully applied to a bioequivalence study of 6 mg of doxepin orally disintegrating tablet formulation in 41 healthy subjects under fasting and fed conditions.
2.Experimental
2.1.Chemicals and materials
Reference standards of Dox hydrochloride(99.8%)and NDox(99.2%)were procured from Toronto Research Chemicals Inc.(Ontario,Canada),while internal standards(ISs),propranolol(99.1%)and desipramine(98.9%)were purchased from Vivan Life Sciences Pvt.Ltd.(Mumbai,India).HPLC grade methanol and acetonitrile,analytical grade ammonia,ammonium acetate and ammonium formate were obtained from S.D.Fine Chemicals Ltd.(Mumbai,India).HPLC grade methyl tert-butyl ether(MTBE)was obtained from J.T Baker Chemicals Ltd.(Haryana,India).Deionized water used for LC–MS/MS was prepared using Milli Q water purification system from Millipore(Bangalore,India).Control buffered(K2EDTA)human plasma was procured from Clinical Department,BA Research India Limited(Ahmedabad,India)and stored at–20 °C until use.
2.2.LC–MS/MS instrumentation and conditions
The liquid chromatography system from Shimadzu(Kyoto,Japan)consisted of an LC-10ADvp pump,an auto sampler(SIL-HTc)and an on-line degasser(DGU-14A).Chromatographic column used was Hypurity C8(100 mm × 4.6 mm,5.0 μm)from Thermo Fisher Scientific Inc.(Waltham,MA,USA).Separation of analytes and ISs were performed under isocratic conditions using a mobile phase consisting of acetonitrile-methanol(95:5,v/v)and 2.0 mM ammonium formate in 93:7(v/v)ratio,delivered at a flow rate of 1.2 mL/min.The auto-sampler temperature was maintained at 4°C and the injection volume was 15 μL.Detection of analytes and ISs was performed on a triple quadrupole mass spectrometer,API-5500 equipped with Turbo Ion spray®,from MDS SCIEX(Toronto,Canada)in the positive ionization mode.Quantitation was done in multiple reaction monitoring(MRM)mode to monitor protonated precursor→ product ion transition of m/z 280.1→107.0,266.0→107.0,260.1→116.1 and 267.1→72.1 for Dox,NDox,propranolol and desipramine,respectively.All the parameters of LC and MS were controlled by Analyst software version 1.5.1.
The source dependent mass parameters maintained for the analytes and ISs were Gas 1(nebulizer gas):50 psi,Gas 2(heater gas):60 psi,ion spray voltage:5500 V,turbo heater temperature:500°C,entrance potential:10 V,collision activation dissociation(CAD):7,curtain gas:30 psi.The compound dependent parameters,namely declustering potential,collision energy and cell exit potential,were set at 60 V,27 eV and 11 V for Dox,60 V,29 eV and 11 V for NDox,81 V,25 eV and 6 V for propranolol and 26 V,21 eV and 10 V for desipramine respectively.Quadrupole 1 and quadrupole 3 were maintained at unit resolution and the dwell time was set at 300 ms.
2.3.Preparation of standard stock and plasma samples
The calibration standards(CSs)were made at 15.0,30.0,60.0,150,300,750,1500,2400,3150 and 3900 pg/mL for Dox and 5.00,10.0,20.0,50.0,100,250,500,800,1050 and 1300 pg/mL for NDox.Quality control(QC)samples(LLOQ QC,lower limit of quantitation quality control;LQC,low quality control;MQC-1&MQC-2,medium quality control;HQC,high quality control;ULOQ QC,upper limit of quantitation quality control)were prepared at 15.0/5.00 pg/mL(LLOQ),45.0/15.0 pg/mL(LQC),360/120 pg/mL(MQC-2),900/300 pg/mL (MQC-1),3000/1000 pg/mL (HQC) and 3900/1300 pg/mL(ULOQ)for Dox/NDox,respectively.
2.4.Protocol for sample preparation
Prior to analysis,spiked plasma/subject samples were thawed and allowed to equilibrate at room temperature.The samples were adequately vortexed using a vortex mixer before pipetting.Aliquots of 500 μL plasma solutions containing 25 μL of combined working solution of Dox and NDox and 475 μL blank plasma were transferred into glass screw tubes.To which,25 μL of methanol:deionized water(50:50,v/v),50 μL combined working solution of ISs was added and vortexed to mix.Further,200 μL of 100 mM ammonium acetate solution(pH 8 adjusted with ammonia)was added and vortexed again.LLE was carried out using 4.0 mL of MTBE by centrifuging the samples for 5.0 min at 1811g.After freezing the aqueous layer in dry ice bath,the organic layer was transferred in clean pre-labeled glass tubes.The samples were then evaporated to dryness at 40°C under gentle stream of nitrogen.The dried samples were reconstituted with 300 μL of acetonitrile:methanol:2.0 mM ammonium formate(80:10:10,v/v/v)and 15 μL was used for injection in LC–MS/MS system.
2.5.Methodology for validation
Method validation for Dox and NDox in human plasma was done following the USFDA guidelines[26]and the procedures followed were similar to our previous work[27].The method was validated for selectivity,interference check,carryover,linearity,precision and accuracy,reinjection reproducibility,recovery,ion suppression/enhancement,matrix effect,stability,dilution integrity and ruggedness.The details are described in Supplementary Material.
2.6.Bioequivalence study design,statistical analysis and incurred sample reanalysis(ISR)
The study design comprised an open label,randomized,two period,two treatment,two sequence,crossover,balanced,single dose,evaluation of relative oral bioavailability of test(6 mg doxepin hydrochloride orally disintegrating tablet from an Indian company)and reference(SILENOR™,6 mg doxepin orally disintegrating tablet from Somaxon Pharmaceuticals Inc.,San Diego,USA)formulations in 41 healthy Indian male subjects under fasting and fed conditions.The procedures followed while dealing with human subjects were based on International Conference on Harmonization,E6 Good Clinical Practice guidelines[28].An ISR was also conducted by computerized selection of 264 subject samples near Cmaxand the elimination phase for both the studies as reported previously[29].The experimental details for both the studies along with statistical analysis are given in Supplementary Material.
3.Results and discussion
3.1.Method development
The objective of the present work was to develop and validate a simple,selective and sensitive method for Dox and NDox in human plasma by LC–MS/MS for routine sample analysis.Additionally,the sensitivity of the method should be adequate to monitor at least five half lives of Dox and NDox concentration with good accuracy and precision for subject samples analysis.During method development,the electrospray ionization of the analytes and ISs was conducted in positive ionization mode using 5.0 ng/mL tuning solution as the drug and its metabolite are basic in nature due to the presence of tertiary and secondary amino groups,respectively.The analytes and ISs gave predominant singly charged protonated precursor[M+H]+ions at m/z 280.1,266.0,260.1 and 267.1 for Dox,NDox,propranolol and desipramine,respectively in Q1 full scan spectra.Further,fragmentation was initiated using sufficient nitrogen for CAD and by applying 30.0 psi curtain gas to break the precursor ions.The most abundant and consistent product ions in Q3 mass spectra of Dox and NDox were found at m/z 107.0,which corresponds to a highly stable hydroxy tropylium ion(Figs.S1 and S2).For propranolol and desipramine,the most stable and reproducible product ions were observed at m/z 116.1 and m/z 72.1,respectively.To reach an ideal Taylor cone for better spectral response,nebulizer gas pressure was set at 50 psi.Fine tuning of nebulizer gas and CAD gas was done to get a consistent and stable response.Ion spray voltage and turbo heater temperature did not have any significant impact on the analyte response and hence were maintained at 5500 V and 500°C,respectively.A dwell time of 300 ms was found adequate for the quantitation of analytes and ISs.Further,no cross talk was observed between the MRMs of the analytes having identical product ions.
The chromatographic conditions were set to obtain adequate separation and resolution of analytes from the endogenous peaks.This included optimization of mobile phase,its composition, flow rate,column type and injection volume.Different combinations of acetonitrile/methanol and acidic buffers(ammonium formate/formic acid,ammonium acetate/acetic acid)of different strengths(2.0–10 mM)were tested as mobile phase.Further,mobile phase additives like formic acid and ammonium trifluoroacetate were also tried on Hypurity C18(100 mm × 4.6 mm,5 μm),ACE C18(100 mm × 4.6 mm,5 μm),Beta Hypersil C18(150 mm × 4.6 mm,5 μm)and Hypurity C8(100 mm × 4.6 mm,5 μm)columns.In addition,the effect of total flow rate was also studied from 0.5 to 1.2 mL/min,which was responsible for acceptable chromatographic peak shapes and separating endogenous peaks.All C18columnsprovided accept ables eparation of analytes with in 3.0 min but there was significant interference of endogenous components,especially with Dox which eluted within 2.0 min.Additionally,the response was inconsistent at LLOQ,CS-1(5.0 pg/mL),CS-2(10.0 pg/mL)and LQC levels for NDox with a small peak tailing.This problem was overcome by using Hypurity C8column,which helped in complete separation of endogenous matrix from the analyte peaks and also provided adequate response at lower concentration levels using a mixture of acetonitrile-methanol(95:5,v/v)and 2.0 mM ammonium formate in 93:7(v/v)ratio as the mobile phase.Addition of 2.0 mM ammonium formate was sufficient to get adequate response and also good peak shape for both the analytes.A flow rate of 1.2 mL/min ensured acceptable peaks shapes with baseline separation of analytes(resolution factor,Rs2.45)within 5.0 min.The retention time for Dox,NDox,propranolol and desipramine were 2.87,3.56,3.15 and 3.77 min,respectively(Figs.1 and 2).The maximum on-column loading(at ULOQ)of Dox and NDox per injection was 97.5 pg and 32.5 pg,respectively.The reproducibility in the measurement of retention time for Dox and NDox was≤1.3%(%CV)for 100 injections on the same column.Due to unavailability of deuterated analogues,some general ISs like desipramine,propranolol,imipramineand amitripty line having structural similarity with the analytes were tested.Based on similar extraction efficiency and chromatographic behavior,propranolol,a beta-blocker,was selected as the IS for Dox and desipramine,a tricyclic antidepressant drug,was preferred as the IS for NDox.Further,both ISs did not affect analytes recovery,sensitivity or ion suppression.
Due to lipophilic nature of Dox and NDox,a vast majority of published methods have used LLE for their isolation from biological matrices with diethyl ether-ethyl acetate[4],n-pentane-isopropanol [11], hexane-propan-2-ol/hexane-dichloromethane mixtures[12],and isoamyl alcohol in hexane[13].In the present work,solvents like n-hexane,MTBE,dichloromethane and ethyl acetate,and their binary mixtures were used to set the optimum conditions for extraction under neutral as well as alkaline conditions.In all the solvent systems,the recovery of Dox and NDox was in the range of 62%–78%and 73%–86%,respectively under neutral conditions.However,the best recovery for both the analytes was found under mild alkaline conditions(pH 8)using MTBE.The recovery obtained was highly consistent and quantitative across all QC levels.
3.2.Assay performance and validation
3.2.1.Selectivity,carryover and interference study
The purpose of evaluating selectivity with 20 different human plasma sources was to determine the extent to which endogenous plasma components might interfere at the retention time of analytes and the ISs and thus,ensure the authenticity of the results for study sample analysis.Figs.1 and 2 demonstrate the selectivity of the method with the chromatograms of double blank plasma,blank plasma spiked with IS,Dox and NDox at LLOQ concentration,respectively and subject samples.Carry-over evaluation was performed in each analytical run to ensure that it does not impact the accuracy and the precision of the proposed method.The experiments showed a carryover of≤0.35%for Dox and≤0.25%for NDox of LLOQ concentration in blank plasma sample after injection of the highest calibration standard(ULOQ)at the retention time of analytes and ISs.Further,there was no interference of commonly used medications by healthy volunteers like acetaminophen,aspirin,caffeine,chlorpheniramine,cetrizine,ibuprofen and pseudoephedrine at the retention time of the analytes and ISs.
3.2.2.Linearity,sensitivity,accuracy and precision
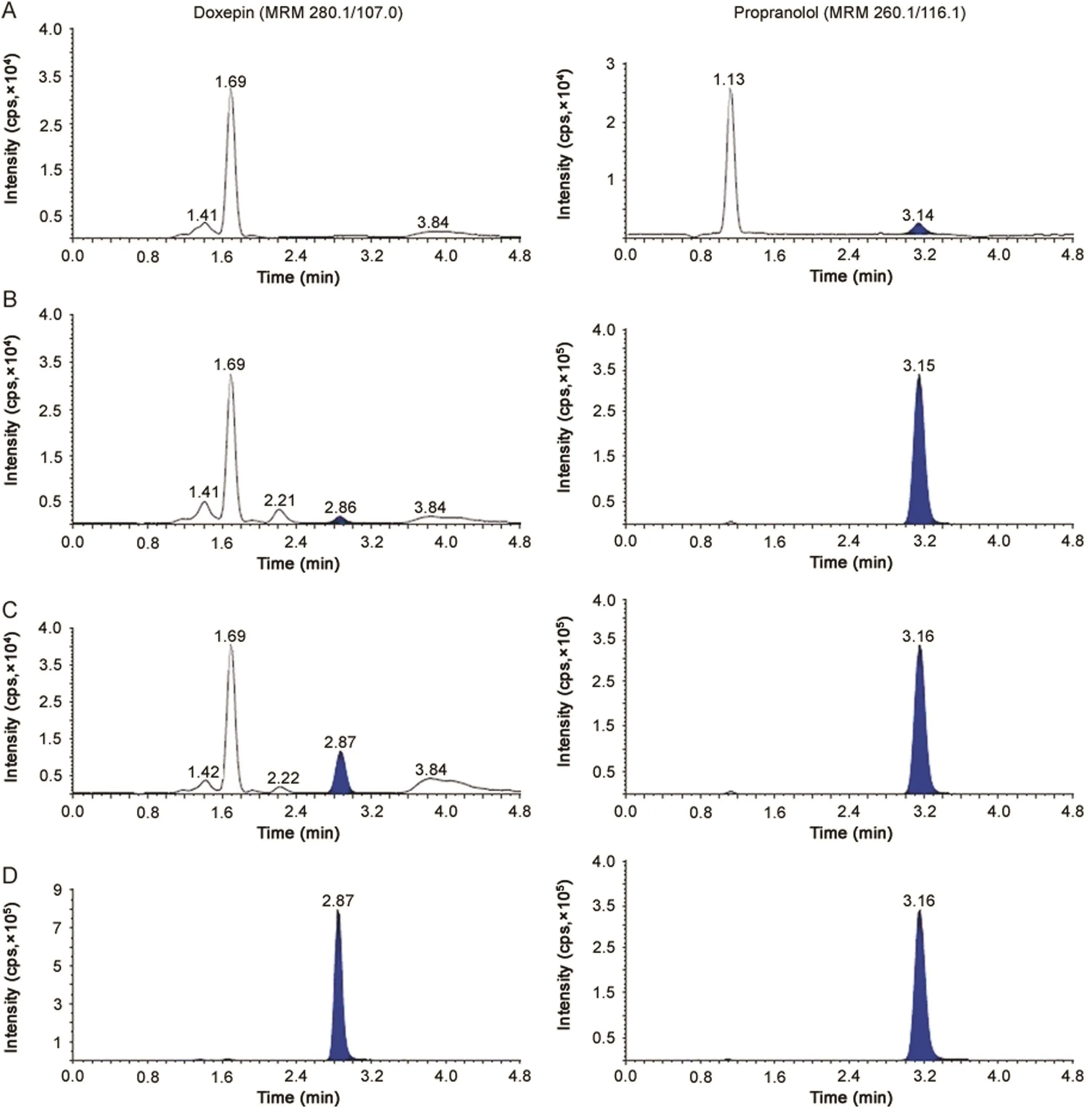
Fig.1.MRM ion-chromatograms of doxepin(m/z 280.1→107.0)and propranolol(IS,m/z 260.1→116.1)in(A)double blank plasma(without analyte and IS),(B)blank plasma with IS,(C)doxepin at LLOQ and IS and(D)real subject sample at Cmaxafter administration of 6 mg dose of doxepin.
The calibration curves for Dox and NDox were linear over the established concentration range of 15.0–3900 pg/mL and 5.00–1300 pg/mL with correlation coefficient(r2)≥ 0.9991 and ≥0.9993,respectively.The mean linear equations computed by least square regression analysis for DOX and NDoxwerey=(0.00192±0.00016)x + (0.00017±0.00003) and y =(0.00117±0.00021)x+(0.00026±0.00002),respectively,where y is the peak area ratio of the analyte/IS and x the concentration of the analyte.The accuracy and precision(%CV)observed for the CSs ranged from 94.4%to 104%and 0.8%–3.7%,respectively for Dox and 95.9%–102%and 1.1%–5.4%,respectively for NDox.The lowest concentration in the standard curves for Dox and NDox was 15.0 pg/mL and 5.00 pg/mL,respectively at a signal-to-noise ratio(S/N)of≥15.
The intra-batch and inter-batch precision and accuracy results are summarized in Table 1.The intra-batch precision(%CV)and accuracy ranged from 1.0%to 8.3%and 93.1%–104.0%,respectively for both the analytes.Similarly for inter-batch experiments,the precision and accuracy varied from 3.4%to 7.2%and 91.7%–101.0%for Dox and NDox.
3.2.3.Recovery,matrix effect and post-column analyte infusion
The extraction recovery and matrix effect data for the analytes and ISs are presented in Table 2.Highly consistent recovery was obtained across QC levels for both the analytes.The IS-normalized matrix factors ranged from 1.02 to 1.05,which shows minimal interference of endogenous matrix components for Dox and NDox.Matrix effect in different plasma sources(6-K2EDTA,1-lipemic and 1-heamolyzed)was also evaluated at LQC and HQC levels.The precision values in different plasma sources varied from 0.3%to 3.9%for both the analytes(Table S1).Further,results of post-column analyte infusion experiment showed no regions of ion suppression or enhancement at the retention time of analytes and IS.
3.2.4.Stability,dilution integrity and ruggedness
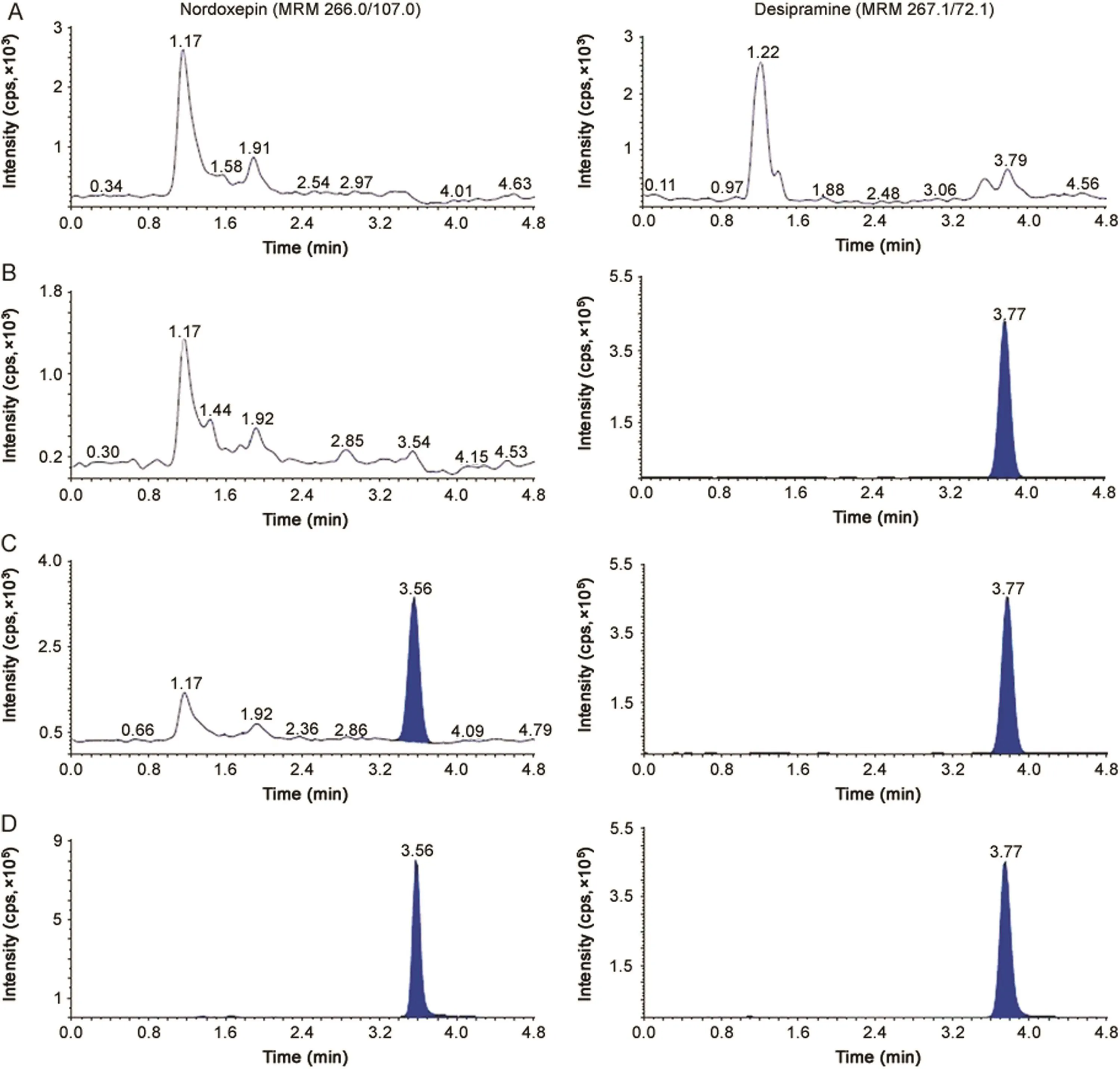
Fig.2.MRM ion-chromatograms of nordoxepin(m/z 266.0→107.0)and desipramine(IS,m/z 267.1→72.1)in(A)double blank plasma(without analyte and IS),(B)blank plasma with IS,(C)nordoxepin at LLOQ and IS and(D)real subject sample at Cmaxafter administration of 6 mg dose of doxepin.
Stability experiments were performed to evaluate the analytes stability in stocks solutions and in plasma samples under different conditions,simulating the same conditions which occurred during sample analysis.Dox and NDox were found stable in controlled blank plasma at room temperature up to 24 h and for six freeze thaw cycles.The analytes in extracted plasma samples were stable for 91 h under refrigerated conditions(2–8 °C)and for 48 h at room temperature.The spiked plasma samples of Dox and NDox stored at-20°C for long-term stability showed evidence of degradation up to 123 days.The detailed stability results are shown in Table 3.
Dilution integrity of the method was checked to confirm dilution reliability of samples having concentration above ULOQ.The precision(%CV)value for 10-fold dilution of 19,500/6500 pg/mL for Dox/NDox was in the range of 2.2%-5.5%and the accuracy results were within 94.4%-102.0%.The results obtained were well within the acceptance limit of 15%for precision(%CV)and 85%–115%for accuracy.Similarly,the precision and accuracy for method ruggedness on two different Hypurity C8columns and with different analysts varied from 1.5%to 6.2%and 93.5%–103.0%,respectively for both the drugs.
3.3.Application of the method in healthy subjects
The validated method was applied to a bioequivalence study of Dox and NDox in 41 healthy Indian males who received 6 mg of test and reference formulations of Dox under fasting and fed conditions.The study was performed to evaluate the impact of food on the pharmacokinetics of Dox and NDox.The method was sensitive enough to monitor their plasma concentration up to 120 h.Fig.3 shows the plasma concentration vs.time profile of Dox and NDox in healthy subjects under fasting and fed conditions.Table 4 gives a comparative assessment of pharmacokinetic parameters obtained for both the studies.As evident from the results,there was minimal effect of food on the pharmacokinetics of Dox and NDox.It was not possible to compare the results obtained with reported methods[11,13]due to limited information on the pharmacokinetic studies of Dox and NDox in healthy subjects.The equivalence statistics of bioavailability for the pharmacokinetic parameters of the two formulations are summarized in Table S2.No statistically significant differences were found between the two formulations in any parameter.Approximately 5060 samples including the calibration,QC and volunteer samples were run and analyzed successfully.The%change in the measurement of selected subject samples for ISR was within±16%,which confirms method reproducibility.
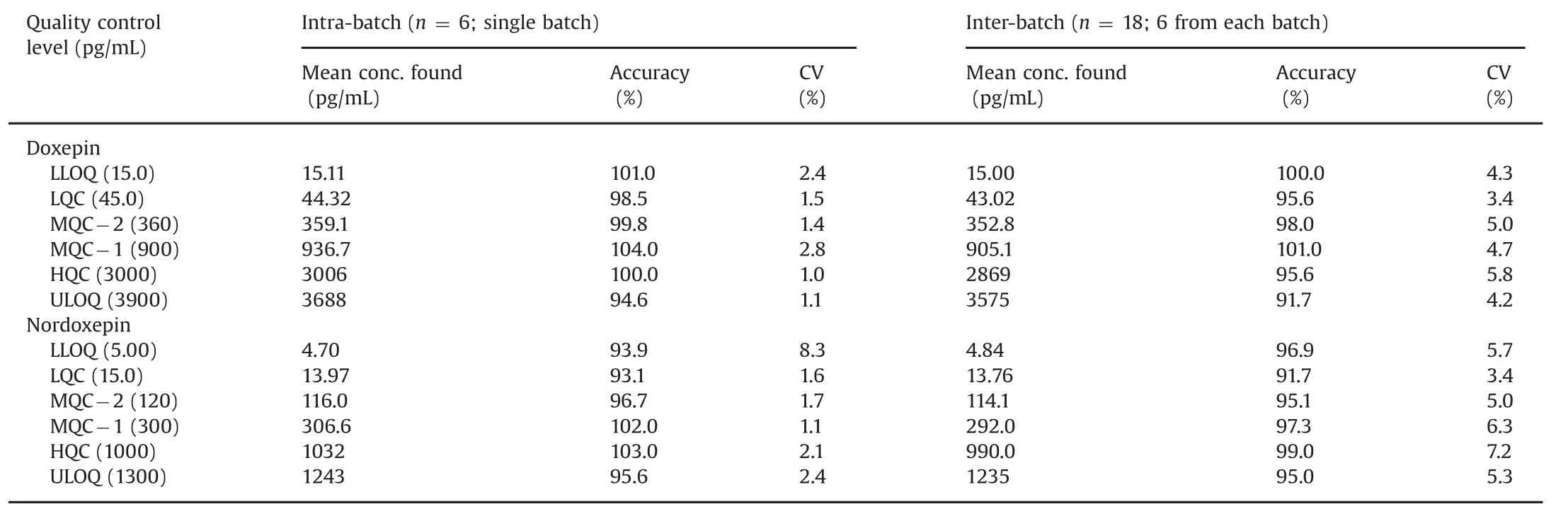
Table 1 Intra-batch and inter-batch accuracy and precision for doxepin and nordoxepin.
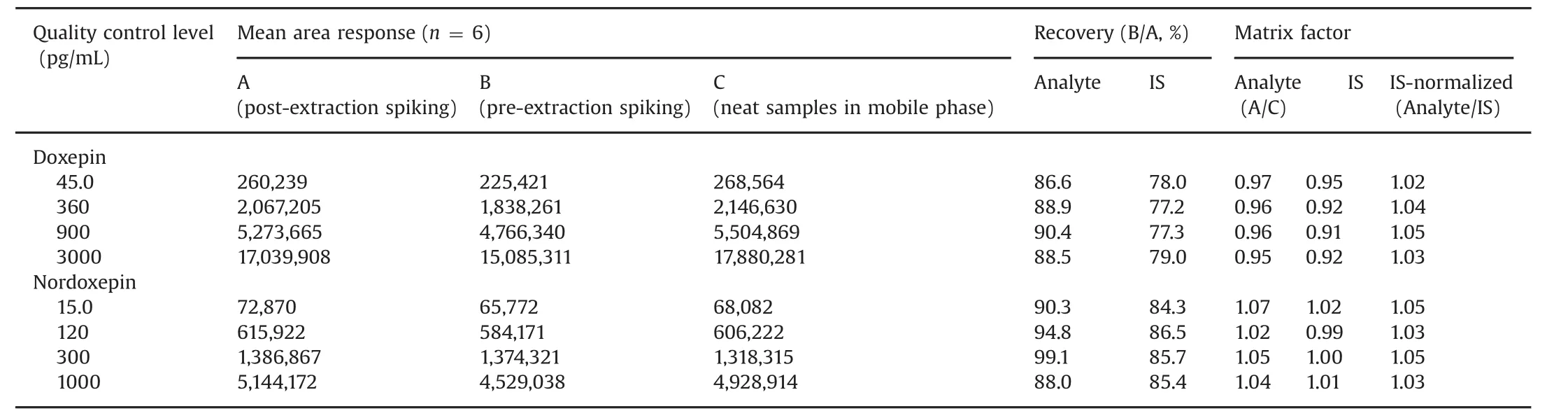
Table 2 Extraction recovery and matrix factor for doxepin and nordoxepin(n=6).
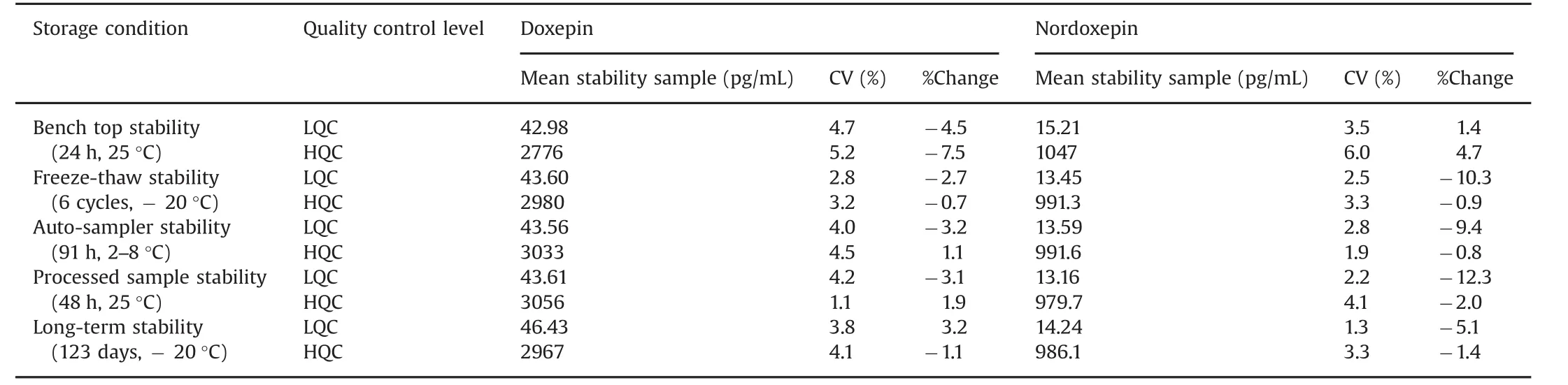
Table 3 Stability results for doxepin and nordoxepin under different conditions(n=6).
4.Conclusion
The present study describes a new,highly sensitive and rugged LC–MS/MS method for the simultaneous determination of Dox and NDox in human plasma,especially to meet the requirement for subject sample analysis.The LLE procedure established using MTBE gave consistent and reproducible recoveries for both the analytes.The optimized linear concentration range was adequate to monitor at least five half-lives of Dox and NDox with good accuracy and precision.The results of clinical study showed no major impact of food on the pharmacokinetics of Dox and NDox.Finally,the reproducibility of the method was adequately proved through incurred sample reanalysis of subject samples for the first time.
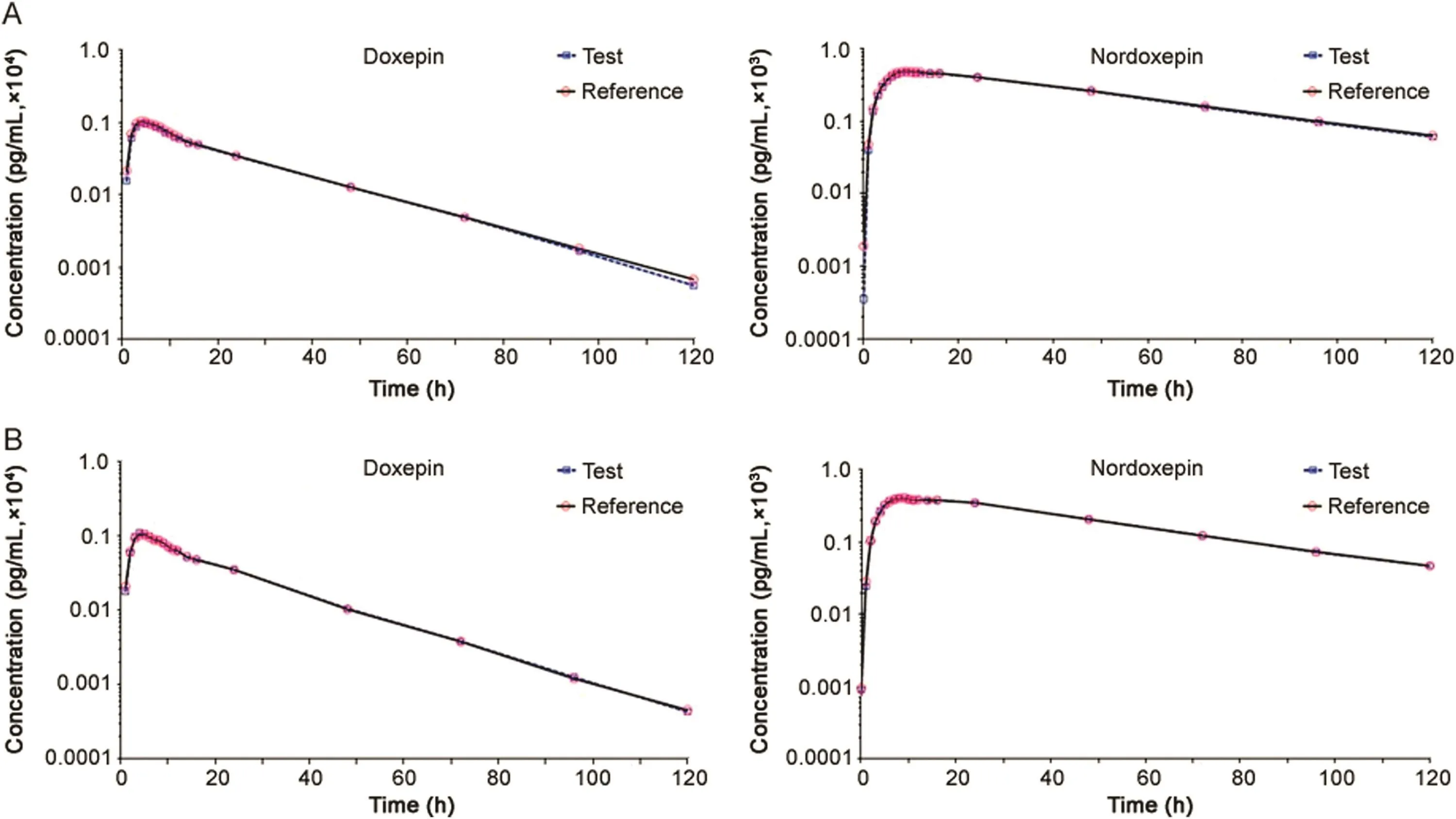
Fig.3.Mean plasma concentration-time profile of doxepin and nordoxepin after oral administration of 6 mg doxepin orally disintegrating tablet(test and reference)formulation to 41 healthy Indian male subjects under(A)fasting and(B)fed conditions.
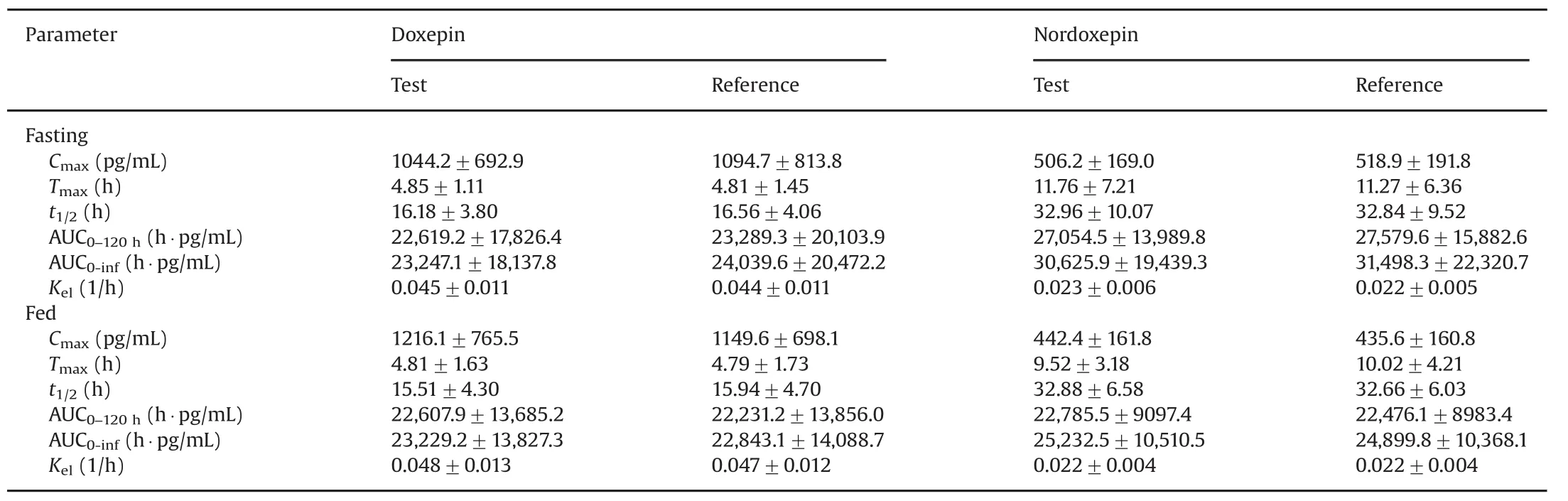
Table 4 Mean pharmacokinetic parameters(±SD)following oral administration of 6 mg of doxepin hydrochloride tablet formulation on 41 healthy Indian male subjects under fasting and fed condition.
Conflicts of interest
The authors have declared that there are no conflicts of interest.
Acknowledgments
The authors are indebted to Mr.Vijay Patel,Executive Director,Cliantha Research Ltd.,for providing necessary facilities to carry out this work and to Mr.Anshul Dogra,Head of Department,Cliantha Research Ltd.,for his support during the course of this project.
Appendix A.Supplementary material
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.jpha.2017.06.004.
杂志排行

Journal of Pharmaceutical Analysis的其它文章
- Novel ligand-based docking;molecular dynamic simulations;and absorption,distribution,metabolism,and excretion approach to analyzing potential acetylcholinesterase inhibitors for Alzheimer's disease
- Recovery rates of combination antibiotic therapy using in vitro microdialysis simulating in vivo conditions
- Evaluation of naproxen-induced oxidative stress,hepatotoxicity and in-vivo genotoxicity in male Wistar rats
- Cytotoxic effect of Rosa canina extract on human colon cancer cells through repression of telomerase expression
- Long-term stability of gentamicin sulfate-ethylenediaminetetraacetic acid disodium salt(EDTA-Na2)solution for catheter locks
- Detection and determination of undeclared synthetic caffeine in weight loss formulations using HPLC-DAD and UHPLC-MS/MS