过硫酸盐微胶囊缓释材料合成及其性能研究
2018-12-06谢先军
敖 琢,徐 瑞,谢先军
(1.湘潭电机股份有限公司,湖南 湘潭 411101;2.中国地质大学(武汉)环境学院,湖北 武汉 430074)
原生高砷地下水已经成为公众饮水安全的严重威胁[1-2]。开发经济高效的高砷地下水水质改良新技术与方法是保障供水水质安全的有效途径。当前,已发展了诸多高砷地下水原位与异位处理方法,并得到了应用[3-6]。与传统的抽出处理技术相比,地下水污染原位处理技术在实际应用中由于具有成本低,且处理彻底、时间短等优势,已成为地下水污染修复技术的主流技术,得到了广泛的关注与应用[7]。目前主要用于地下水污染原位处理的方法有生物修复、反应性渗透墙(PRB)、曝气除砷等技术,其中反应性渗透墙被认为是最有效的地下水污染原位去除技术之一,尤其在地下水砷的原位去除中展现出良好的应用前景[8]。但是,PRB技术也具有一些缺陷使其无法很好地适用于原生高砷地下水的修复,如:①原生高砷地下水常以带状或面状大面积分布;②高砷地下水通常赋存于十余米至数十米深的含水层中,PRB技术施工工程量巨大,技术要求高,费用昂贵。
近几年的研究表明,可以通过引入亚铁盐和相应的氧化剂至高砷含水层,促进沉积物表面形成铁(氢)氧化物,并利用铁(氢)氧化物对砷的强烈吸附作用促进砷的固定[9-10]。碱性条件下,二价铁被氧化后在沉积物表面形成水铁矿或针铁矿,可抑制砷的释放[10-11]。然而,高砷地下水通常发生于还原环境中,该条件下铁(氢)氧化物可能发生还原性溶解并导致砷的再次释放。在氧化条件或弱到中等还原条件下,铁(氢)氧化物则能稳定存在一年以上[12]。因此,长效调控地下水的氧化还原环境以促进铁(氢)氧化物的生成是当前研究的一个重要课题。纳米缓释材料是指通过物理、化学等方法改变材料结构,使材料的活性成分在设定的空间及预定的时间内缓慢释放于环境中,并使材料较长时间维持在有效浓度内的一类新型材料。该新型材料与常规材料相比,具有延长持效期、减少材料添加次数、降低分解与流失等优点。因此,利用优良的纳米载体材料和先进的纳米加工技术制备性能优良的纳米缓释材料,将其应用于原位除砷体系,可以有效地改进传统原位除砷处理技术的功能缺陷,改善材料的分散性、释放速度、剂量转移效率和地下水的氧化还原环境,是提高材料有效利用率和固砷效果的可行途径。简言之,纳米缓释技术应用于地下水原位除砷,其目的主要不是引入新的除砷材料体系,而是通过将氧化剂负载到纳米材料载体上,并缓慢释放到地下水环境中,使地下水环境长期处于氧化态,从而使传统的铁基除砷材料长效稳定地发挥作用,并防止因铁基材料的还原性溶解导致已吸附砷的流失,造成二次污染。
微胶囊技术是指利用特定的材料将化学药剂或药品包裹其中,形成直径几千微米以下的的微小胶囊结构的技术,其主要用于药物的缓释与控释过程中[13-15]。本文拟通过原位聚合法制备的聚吡咯包裹过硫酸钾的微胶囊缓释材料,用于在地下水环境中缓慢释放氧化剂,以调节地下水中的氧化还原环境。该微胶囊具有很明显的“核-壳”结构,它由壳材包裹着芯材组成,通过壳材的屏蔽能起到一个保护芯材的作用,使得芯材的性质不发生变化[16]。芯材可以为水溶性、油溶性或者混合物质组成,芯材的状态可以是固态、液态、气态或者粉末等[17]。壳材的范围比较局限,一般根据芯材的性质选取相应的壳材,可用作壳材的材料有天然高分子、聚合物、半合成高分子、合成高分子以及部分金属。水溶性的芯材一般在油性溶剂当中制备,同样油溶性的芯材在水溶液当中制备,这样做的目的主要是使得芯材不被溶解,水溶性的芯材选取油溶性的壳材在油性溶剂当中制备微胶囊,其壳材可能会溶解到油性溶剂当中,通过蒸发、挥发等手段将溶剂除去,即可得到微胶囊[18]。本文的目的是将过硫酸钾微胶囊化,使其缓慢释放,而过硫酸钾是水溶性芯材,故需要在油性溶剂中制备。
1 材料与方法
1. 1 试验原料
试验原料:过硫酸钾、乙酸、十二烷基硫酸钠(SDS)、聚乙二醇6000(PEG6000)、乙醇、丁醇。吡咯、甘油、异丙醇、司盘80(Span 80)等,均为分析纯,且全部购自国药集团化学试剂有限公司。试验用水为去离子水。
1. 2 微胶囊样品的制备
以十二烷基硫酸钠(SDS)、聚乙二醇6000和去离子水的混合液作为A液,异丙醇、司盘80和丁醇的混合液作为B液,将上述A、B液混合后装入容量为100 mL的三口烧瓶内,之后再加入乙醇和乙酸[具体用量:10 mL去离子水,0.2 g十二烷基硫酸钾(SDS),0.32 g聚乙二醇6000,30 mL异丙醇,10 mL丁醇,0.2 mL司盘80,5 g过硫酸钾,2 mL乙醇以及4 mL乙酸];将该混合物搅拌1 h并加入乙酸调节pH值至2;快速滴加丙三醇和吡咯的混合物,在恒定的温度下继续搅拌4~6 h,静置,沉淀,析出上清液;用水和无水乙醇对沉淀物进行洗涤,分别洗涤4~6次,并置于55 ℃条件下真空烘干24 h,即得到微胶囊样品。
1. 3 微胶囊样品的测试与表征
采用场发射扫描电子显微镜(SEM,SU8010)观察微胶囊样品的表面形貌;采用能谱仪(EDS)对微胶囊样品进行成分分析;通过X射线粉末多晶衍射(XRD,D8-Focus)表征微胶囊样品的晶型结构,以Cu K (λ= 1.540 598 Å)为辐射源,扫描范围一般为10°~70°,步长为0.01°,每步扫描时间为0.05 s;利用电导率仪(DDS-11A,China)检测微胶囊样品中过硫酸钾在水中的释放情况。
2 结果与讨论
2. 1 微胶囊的晶体结构分析
由于过硫酸钾属于无机盐,所以它具有很清晰的晶型结构,而聚吡咯属于无晶型结构,可以通过微胶囊样品的X射线衍射(XRD)图谱分析来判断试验中的过硫酸钾是否成功被包裹在聚吡咯之中。图1为微胶囊的XRD图谱,其中黑色部分(上方)为聚吡咯包裹过硫酸钾的XRD图谱,红色部分(下方)为标准过硫酸钾的XRD图谱。
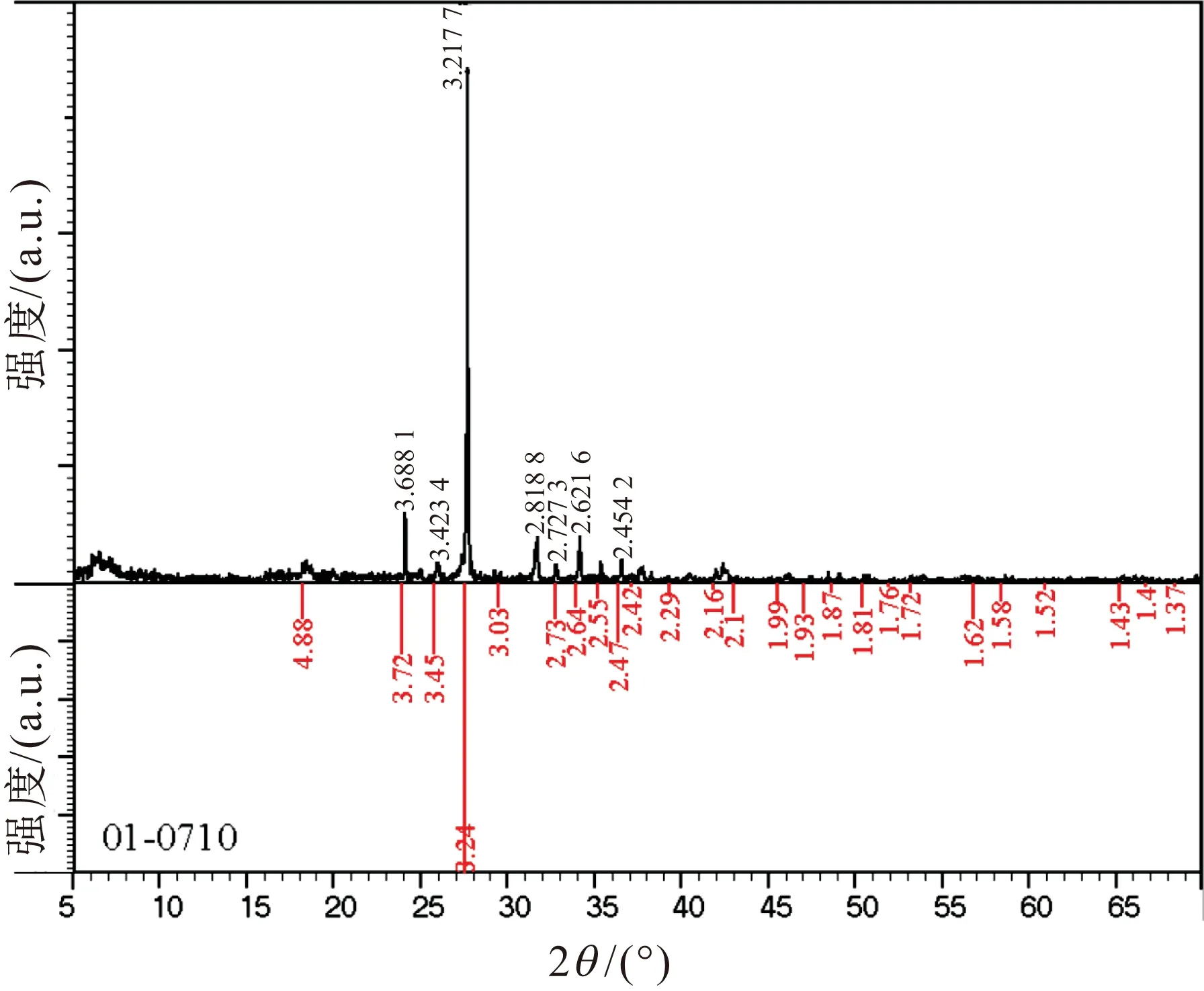
图1 微胶囊的XRD图谱Fig.1 XRD pattern of microcapsule
由图1可见,标准过硫酸钾的XRD图谱中的特征峰(2θ)在50.8°、41.7°、35.4°、27.4°、23.6°、18.5°处,它们分别对应了过硫酸钾(220)、(030)、(012)、(020)、(101)、(110)衍射晶面[45];本试验所制备的微胶囊样品对应的特征峰与标准过硫酸钾的特征峰相吻合,除了峰的强度有细微的变化之外,峰的位置并未发生明显的变化,这也说明过硫酸钾经过原位聚合法微胶囊化后,其晶体结构未发生变化。这是因为微胶囊的壳层聚合物属于无晶型结构,所以在XRD图谱中没有出现其他的特征峰。根据微胶囊样品XRD的表征结果,初步确定过硫酸钾成功地被聚合物包裹。
2. 2 微胶囊的微观形貌及元素分析
图2为微胶囊的扫描电镜图。
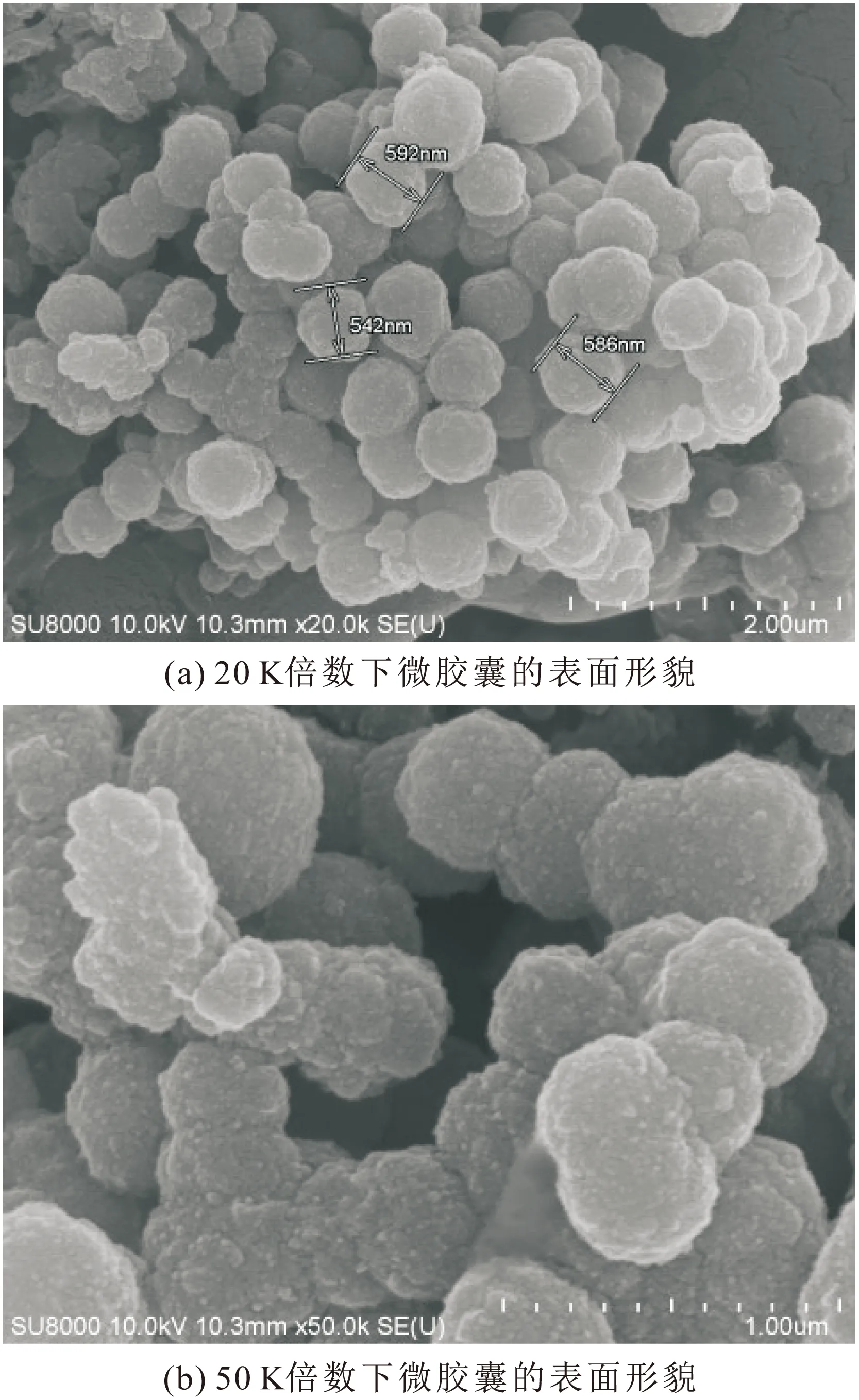
图2 微胶囊的扫描电镜图Fig.2 SEM image of microcapsule
由图2可见,本试验所制备的微胶囊样品是分散的、球状的、直径在500~600 nm的纳米颗粒,进一步证明本试验合成了均匀的聚吡咯包裹过硫酸钾的微胶囊。
图3为微胶囊的能谱图。

图3 微胶囊的能谱图Fig.3 EDS spectra of microcapsule
由图3可见,本试验所制备的微胶囊样品中,O元素的原子数量百分比为27.31%,占样品总质量的31.09%;C元素的原子数量百分比为53.65%,占样品总质量的45.85%;N元素的原子数量百分比为15.97%,占样品总质量的15.92%;S元素的原子数量百分比为2.72%,占样品总质量的6.20%;K元素的原子数量百分比为0.34%,占样品总质量的0.95%,进一步表明过硫酸钾已经成功被聚合物包裹。
2. 3 微胶囊的形成和释放机理
本试验采用原位聚合法制备过硫酸钾微胶囊,其特点是在微胶囊制备过程中,单体(吡咯)与固体过硫酸钾颗粒处于同一连续相中,并随着反应的进行,在同时为引发剂的过硫酸钾作用下,可溶的吡咯单体逐渐变为不可溶的聚吡咯,并从连续相中析出,包覆在过硫酸钾表面形成“核(过硫酸钾)-壳(聚吡咯)”结构的微胶囊。由于吡咯单体遇到过硫酸钾后会立即发生聚合反应,容易导致所制备的微胶囊互相黏连,从而影响微胶囊的释放性能。因此,在采用原位聚合法制备过硫酸钾微胶囊前首先要考虑的问题是如何选取合适的稳定剂,使聚吡咯能够将过硫酸钾颗粒完整地包裹,并确保形成的微胶囊在稳定剂的作用下具有较好的分散性。Span 80为黄色油状液体,能溶于二氯甲院、三氯甲烷、乙酸乙酯、丙二醇、乙醇、甲醇或液体石蜡等有机溶剂中,其亲水亲油平衡值为4.3,常用作W/O型乳液的乳化剂,也用作微胶囊的分散稳定剂;SDS、PEG6000均为长链阴离子型表面活性剂,其亲水基团能吸附在过硫酸钾颗粒表面,亲油基团伸向油溶性的吡咯单体,使得吡咯单体能较好地在过硫酸钾表面吸附并聚合。可见,在上述这种稳定剂体系中能制备“核-壳”结构的微胶囊,并且所制备的微胶囊具有一定的分散性。如图2所示,复合表面活性剂作为稳定剂体系,能制备过硫酸钾微胶囊,并且微胶囊呈明显的“核-壳”结构,微胶囊的分散性相比单一表面活性剂作为稳定剂体系中制备的微胶囊有一定程度的提高。因此,本文选择Span 80/SDS/PEG6000复合表面活性剂作为制备过硫酸钾微胶囊的稳定剂。
过硫酸钾微胶囊形成的过程主要分为3个步骤:第一步为表面活性剂十二烷基硫酸钠中的亲水基团由于静电吸附的作用,使十二烷基硫酸钠得以固定于过硫酸钾颗粒的表面,比如十二烷基硫酸钠中的亲水基团和过硫酸钾表面对应的硫酸根发生吸附反应;第二步为表面活性剂PEG6000中的部分亲水基团伸向具有水溶性的过硫酸钾颗粒的表面,另外一部分基团则吸附十二烷基硫酸钠中的部分亲水基团;第三步为复合表面活性剂(SDS/PEG6000)中的疏水基团伸向油溶性的甘油掺杂之后产生的吡咯单体,使吡咯在过硫酸钾的表面发生积聚,同时在过硫酸钾的引发作用下发生聚合反应,形成微胶囊的壳层。而Span 80使得制备的微胶囊分散性更好。
在本次释放试验中,将制备的微胶囊样品置于不同条件下的水体系,水便会从聚吡咯的丙三醇中渗透进入微胶囊的内部,进而发生反应。首先,水由丙三醇分子中进入壳层的聚吡咯中,进而使过硫酸钾发生溶解,这时由于微胶囊内部和外部的水介质中存在一定的浓度差,过硫酸钾便可以根据扩散作用而进入到聚吡咯的表面;然后由于过硫酸钾的扩散作用而进入到外部的水介质当中。本释放试验中通过采用电导率仪来监测不同甘油条件下对应的过硫酸钾释放速率和释放时间。
2. 4 微胶囊的缓释性能
本次释放试验主要测试了丙三醇(甘油)用量对过硫酸钾微胶囊释放时间的影响,即主要通过改变制备过硫酸钾微胶囊过程中的丙三醇用量来对制备的过硫酸钾微胶囊样品的完全释放时间进行测试,并判断制备过程中丙三醇用量对制备的微胶囊样品缓释性能的影响情况。具体方法是:在微胶囊样品的制备过程中,改变丙三醇用量(分别添加0.4 mL、 0.6 mL、0.8 mL)来研究微胶囊样品的完全释放时间;之后,在pH值为7.0、NaCl浓度为100 mg/L、温度为28℃的条件下,通过对微胶囊样品释放过程中溶液电导率的测量,计算出微胶囊相对释放量的百分比,进而分析丙三醇的添加量对微胶囊缓释性能的影响。图4为不同甘油添加量条件下微胶囊的释放情况。

图4 不同甘油添加量条件下微胶囊的释放情况Fig.4 Release amounts of microcapsules under different glycerol addition conditions
由图4可见,当丙三醇的添加量为0.4 mL时,包裹在微胶囊中的过硫酸钾几乎无法释放;当丙三醇的添加量分别为0.6 mL和0.8 mL时,包裹在微胶囊中的过硫酸钾有大量释放。其中,当丙三醇的添加量为0.6 mL时,微胶囊的相对释放量百分比大约在13 h达到最大值;当丙三醇的添加量为0.8 mL时,微胶囊的相对释放量百分比大约在10 h达到最大值;随着丙三醇添加量的增加,微胶囊的起始释放时间不断缩短,达到相对释放量最大值的时间也缩短。分析原因认为:共聚物壳中的丙三醇在水体系中会发生溶胀现象,溶胀现象是引起包裹在微胶囊中的过硫酸钾释放的主要原因,而丙三醇的添加量增大会使水更容易地由丙三醇分子进入壳层的聚吡咯中,进而使过硫酸钾发生溶解;过硫酸钾的逐渐溶解又会导致微胶囊内部和外部的浓度差逐渐增大,随后过硫酸钾便根据扩散作用更多地进入到聚吡咯的表面,紧接着逐渐进入到外部的水介质当中。
通过测试丙三醇的用量对微胶囊释放过硫酸钾速率的影响试验可知:当丙三醇的用量降低时,微胶囊壳层中的丙三醇比例减少,使得在释放过程中水进入微胶囊内部的通道变少,从而减缓过硫酸钾的释放速率;但当丙三醇的用量降低到一定值时,微胶囊壳层中丙三醇可能全部被聚吡咯包裹,使得水无法通过聚吡咯接触丙三醇,从而完全不能进入微胶囊内部,此时整个微胶囊是被聚吡咯完全封闭的,过硫酸钾完全不能释放出来。
3 结论与建议
本文通过原位聚合法成功制备了以过硫酸钾为核、聚吡咯为壳的微胶囊缓释材料即微胶囊样品,其最长释放时间能达到14 h。通过X射线粉末多晶衍射、场发射扫描电镜、能谱仪等表征手段考察了所制备的微胶囊样品的组成、表面形貌,并利用电导率仪检测了微胶囊样品中过硫酸盐在水中的释放情况。试验结果表明:聚吡咯成功地将过硫酸钾包裹,且没有改变过硫酸钾的晶体结构;微胶囊样品中的芯材过硫酸钾可在水体系中完全释放出来,并且其释放时间和释放速率可通过调节微胶囊的壳材组成进行调控。后续研究可考虑对材料表面改性、接枝疏水基团等方式来延缓其释放速率。原位聚合法制备微胶囊在高砷地下水环境中可以起到长效调控氧化还原环境的作用,使得修复系统中固砷的铁(氢)氧化合物长期稳定存在,从而提高高砷地下水原位修复的效率。
