小反刍兽疫病毒实时荧光定量PCR检测方法的建立
2018-12-05付明哲郝玉青许信刚
付明哲,杨 峰,郝玉青,许信刚,张 琪*
(1.西北农林科技大学动物医学院,陕西杨凌 712100;2.榆林市动物疫病预防控制中心,陕西榆林 719000)
小反刍兽疫病毒(Peste des petits ruminants virus,PRRV)属于副黏病毒科麻疹病毒属,羊、鹿等小型反刍类动物感染PPRV后导致以稽留热、坏死性胃肠炎和严重腹泻为主要临床症状的急性接触性传染病小反刍兽疫,我国将该病列为一类动物疫病[1-2]。山羊和绵羊对PPRV最为易感,其感染后的发病率和病死率可高达100%。我国自2007年暴发小反刍兽疫,随后全国23个省、市、自治区地区都有该病的报道,近年呈区域内点状散发,给我国畜牧业的发展造成了困扰[3-5],因此,一种特异、灵敏、量化且耗时短的检测PPRV的方法对小反刍兽疫的防控很有必要。目前实验室检测PPRV的最常用方法是反转录PCR(reverse transcription PCR,RT-PCR)方法,然而在灵敏度和病毒含量测定以及检测速度等方面尚存在一些不足[6]。实时荧光定量PCR(real-time fluorescence quantitative PCR,qPCR)凭借其简单的操作,较高的灵敏度,良好的重复性和量化的结果以及较短的检测时耗等优点[7],逐渐在实验室的检测中得到了广泛的应用,尤其是费用相对较低的荧光染料法实时定量PCR[8-10]。本研究根据NCBI中登录的PPRV的N基因设计特异性引物,建立基于Eva Green的反转录实时荧光定量PCR(RT-qPCR)检测方法,以期为PPRV的临床检测提供一种特异、灵敏、量化及快速的方法。
1 材料与方法
1.1 材料
1.1.1 毒株、菌株 小反刍兽疫病毒疫苗购自新疆天康畜牧生物技术股份有限公司;羊口疮疫苗购自山东泰丰生物有限公司;山羊痘疫苗购自山东绿都公司;羊流产嗜性衣原体由西北农林科技大学动物医学院兽医微生物实验室分离保存;感受态细胞DH5α购自康维世纪生物科技公司。
1.1.2 待检病料 16份临床PPRV cDNA样品由榆林市动物疫病预防控制中心提供,主要是2014年陕北榆林地区由农业部国家外来动物疫病防控制中心确诊的养殖场病料样品提取RNA后逆转录的cDNA,全部病羊按照国家相关要求处理[3,6],确诊为PPRV阴性的脾脏由西北农林科技大学动物医学院微生物实验室提供。
1.1.3 主要试剂 DNA Marker DL 2 000和2×TaqMaster Mix购自康维世纪生物科技公司;Trizol试剂、RNA逆转录试剂盒购自天根生化科技公司;琼脂糖凝胶回收试剂盒、pEAST-T1克隆试剂盒、质粒提取试剂盒购自全式金生物公司;2×Eva Green Premix购自ABM生物科技公司。
1.2 方法
1.2.1 引物设计及合成 通过对比分析NCBI中登录的小反刍兽疫病毒(HQ197753.1)的N基因序列保守序列,用Primer 5设计一对特异性引物。所设计的荧光定量特异性上游和下游引物序列分别为PPRV-F:5′-TCCATCATTACCCGTTCA-3′,PPRV-R:5′-TGTCCAAATCAGCACCAC-3′,扩增片段长度为244 bp。引物由西安擎科泽西生物公司合成,稀释至浓度为10 μmol/L,4℃保存备用。
1.2.2 PPRV RNA的提取及逆转录 取稀释的疫苗病毒液200 μL,加1 mL Trizol试剂,使用Trizol法提RNA,使用逆转录试剂盒进行逆转录,获得的cDNA在-20℃存储或进行后续试验。
1.2.3 常规RT-PCR扩增 50 μL反应体系:2×TaqMaster Mix和模板分别为25 μL、6 μL,上、下游引物均为4 μL,双蒸水补足。反应条件:95℃ 2 min;95℃ 30 s,56℃ 30 s,72℃ 30 s,30次循环;72℃ 2 min。
1.2.4 RT-qPCR阳性质粒及标准模板制备 按照胶回收试剂盒回收1.2.3扩增的目的片段,并将其按照pEAST-T1克隆试剂盒构建重组质粒菌。重组质粒菌经常规PCR鉴定为阳性后,将重组质粒菌中的阳性质粒按照试剂盒的操作步骤提取,进行测序分析。用超微量光谱分析仪测定阳性质粒浓度,按照以下公式将浓度转化为拷贝数,然后进行10-1~10-9的梯度稀释。稀释后的质粒作为标准模板,放置-20℃保存备用。拷贝数(copies/μL)=C×NA/MW,其中C为阳性质粒测定的浓度,单位ng/μL,NA取值为6.02×1023copies/mol,MW为平均分子质量,单位Dolton。
1.2.5 RT-qPCR扩增 10 μL反应体系:2×Eva Green Premix、标准模板、上游和下游引物分别为5 μL、1 μL、0.3 μL、0.3 μL,ddH2O补足。反应条件:95℃ 10 min:95℃ 3 s,60℃ 30 s,采集荧光信号,35次循环。
1.2.6 RT-qPCR反应标准曲线建立 选取浓度稀释10-4~10-8的标准模板为阳性模板,以双蒸水为阴性对照模板,进行RT-qPCR扩增。结果以实时荧光定量PCR仪(TL988)自带软件分析绘制扩增曲线、标准曲线和熔解曲线。
1.2.7 RT-qPCR特异性试验 用建立的RT-qPCR方法分别检测PPRV、羊口疮病毒(ORFV)、山羊痘病毒(GTPV)、羊流产嗜性衣原体的cDNA或者DNA,验证建立的RT-qPCR方法的特异性。
1.2.8 RT-qPCR灵敏性试验 选取浓度稀释了10-6~10-10的标准模板为阳性模板,双蒸水为阴性对照模板,RT-qPCR方法扩增。选取浓度稀释了10-6~10-9的标准模板为阳性模板,双蒸水为阴性对照模板,常规PCR方法扩增。通过比较两种方法可以检测到阳性结果的最低浓度,以此得出灵敏程度。
1.2.9 RT-qPCR重复性试验 分别以同一批次104~106copies/μL浓度的标准模板和不同批次104~106copies/μL浓度的标准模板为阳性模板,用双蒸水作为阴性对照模板,进行RT-qPCR扩增,计算组内和组间变异系数。
1.2.10 临床样品的检测 取16份临床PPRV RNA反转录的cDNA样品做待测样本,以标准模板做阳性对照模板,以双蒸水为阴性对照模板,用所建立的RT-qPCR方法检测。
2 结果
2.1 阳性质粒PCR鉴定
将菌液用荧光定量特异性引物进行PCR检测,结果为244 bp目的片段(图1)。将鉴定为阳性的质粒测序,通过Blast比对,结果同源性达100%,说明重组质粒构建成功。
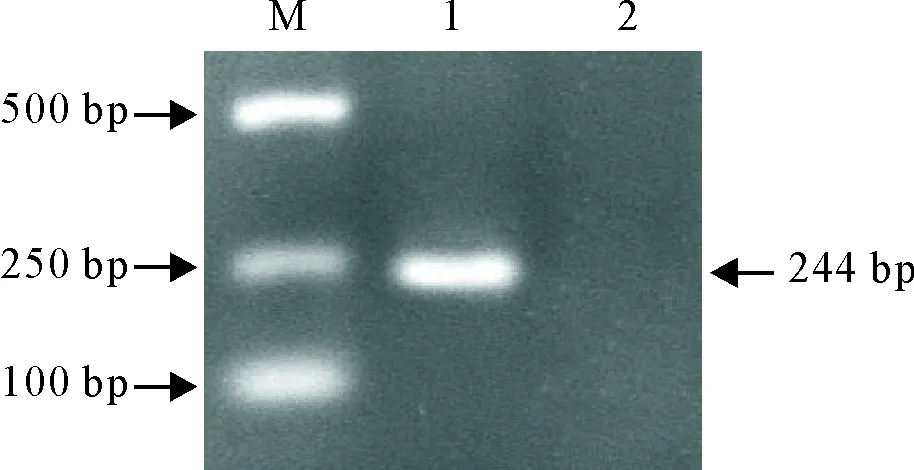
M.DNA标准DL 2 000;1.阳性质粒PCR产物;2.阴性对照
M.DNA Maker DL 2 000; 1.PCR products of positive plasmid; 2.Negative control
图1阳性质粒PCR鉴定
Fig.1 PCR identification of positive plasmid
2.2 RT-qPCR标准模板的制备
用超微量光谱分析仪测定提取质粒的浓度为112.5 ng/μL,OD260/OD280值为1.827,计算得拷贝数为2.46×1010copies/μL,将其稀释至浓度分别为2.46×109copies/μL、2.46×108copies/μL、2.46×107copies/μL、2.46×106copies/μL、2.46×105copies/μL、2.46×104copies/μL、2.46×103copies/μL、2.46×102copies/μL、2.46×101copies/μL、2.46×100copies/μL,置-20℃保存备用。
2.3 RT-qPCR反应标准曲线建立
建立标准模板的RT-qPCR扩增曲线见图2。相应的标准曲线见图3。其熔解曲线见图4。在2.46×102copies/μL~2.46×107copies/μL的浓度区间内,所得拷贝数(x)与Ct值(y)的线性方程为y=-3.39x+37.637,斜率为-3.39,截距为37.637,线性相关系数R2=0.999,显示了良好的线性关系。熔解曲线峰型单一,无杂峰,熔解温度一致,Tm=84.9℃。
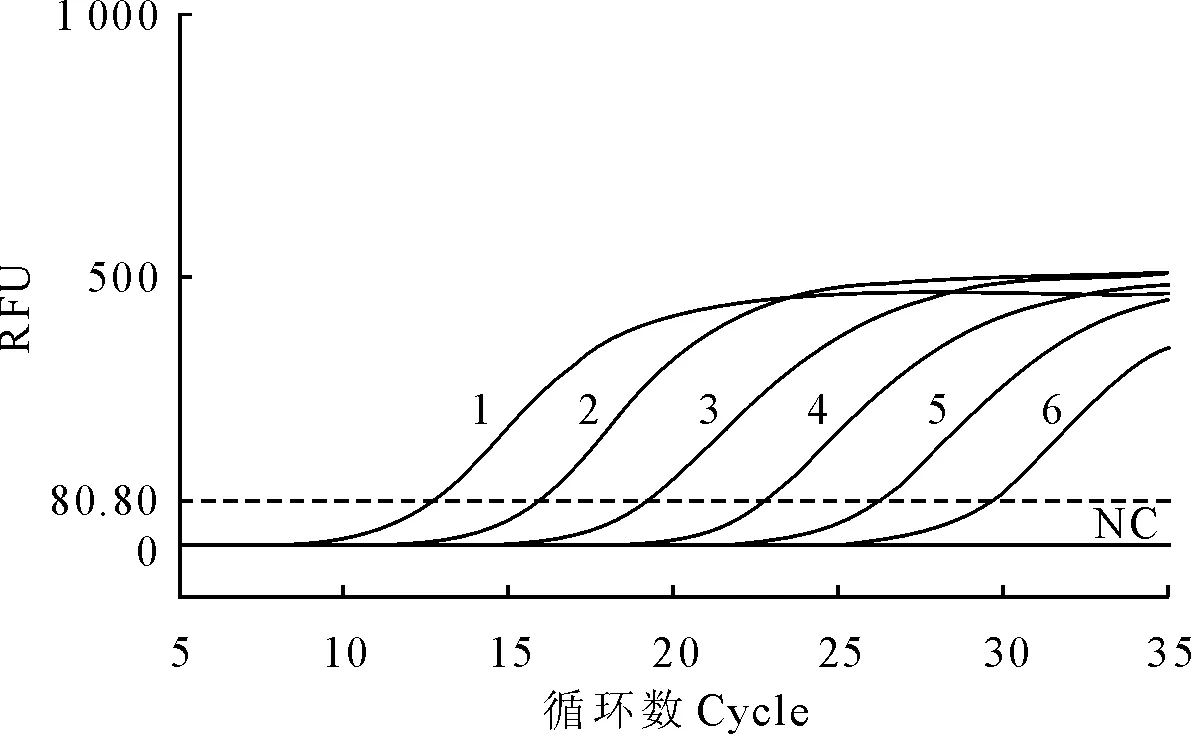
1~6.2.46×107copies/μL~2.466×102copies/μL 6个不同浓度标准模板的qRT-PCR扩增曲线;NC.阴性对照
1-6.qRT-PCR amplification curves of 2.46×107copies/μL to 2.46×102copies/μL diluted standard template,respectively; NC.Negative control
图2实时荧光定量PCR扩增曲线
Fig.2 The amplification curve of RT-qPCR
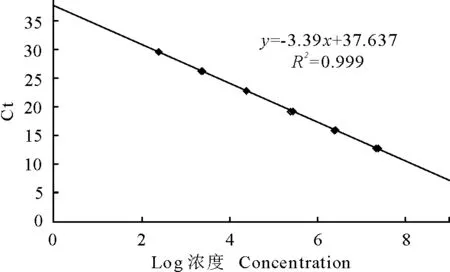
图3 实时荧光定量PCR标准曲线

图4 实时荧光定量PCR熔解曲线
2.4 RT-qPCR特异性试验
分别以PPRV、ORFV、GTPV和羊流产嗜性衣原体以及确诊PPRV阴性的脾脏cDNA或者DNA为模板,用建立的RT-qPCR方法扩增,结果只有PPRV有荧光信号(图5),说明所建立的RT-qPCR方法特异性好。

图5 RT-qPCR特异性试验扩增曲线
2.5 RT-qPCR灵敏性试验
以2.46×104copies/μL~2.46×100copies/μL的标准模板为模板,进行RT-qPCR方法扩增。以2.46×105copies/μL~2.46×101copies/μL的标准模板作为模板,用常规PCR方法扩增。结果表明,RT-qPCR方法检出限为2.46×101copies/μL(图6),常规PCR方法检出限为2.46×103copies/μL(图7),说明建立的RT-qPCR检测方法与常规RT-PCR相比灵敏100倍。

1~5.2.46×104copies/μL~2.46×100copies/μL 5个不同浓度标准模板的RT-qPCR扩增曲线;NC.阴性对照
1-5.RT-qPCR amplification curves of 2.466×104copies/μL to 2.466×100copies/μL diluted standard template,respectively; NC.Negative control
图6 RT-qPCR灵敏性试验
Fig.6 Sensitivity test of RT-qPCR
2.6 RT-qPCR重复性试验
以2.46×106copies/μL、2.46×105copies/μL、2.46×104copies/μL的标准模板为反应模板,每个模板做3个重复,进行组内重复试验(表1),每隔1周做1次,每次做3个重复,计算平均值,做3次,进行组间重复试验(表2)。结果显示,建立的RT-qPCR方法的变异系数在1%以下,其重复性表现优异。
2.7 方法的初步应用
应用本试验所建立的RT-qPCR检测方法对16份临床病料提取RNA并反转录的cDNA样品进行检测,并与RT-PCR作对比。检测结果显示, RT-PCR法检测到阳性样品16份,RT-qPCR方法检测到阳性样品也为16份,RT-qPCR方法与常规RT-PCR方法检测结果一致。
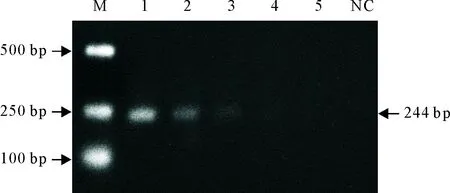
M.DNA标准DL 2 000;1~4.2.46×105copies/μL~2.46×101copies/μL 4个不同浓度标准模板的常规PCR产物;NC.阴性对照
M.DNA Maker DL 2 000; 1-4.The PCR products of 2.46×105copies/μL to 2.46×101copies/μL diluted standard template; NC.Negative control

图7 常规PCR灵敏性试验
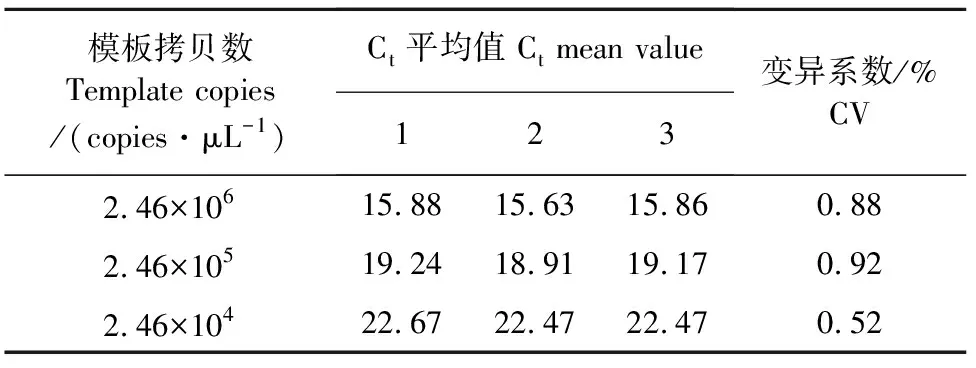
表2 RT-qPCR组间重复试验
3 讨论
我国小反刍兽疫尽管在2015年后已得到控制,但仍呈地方散发,通过实验室检测手段对早期感染和隐性带毒者高效、快速的检出,可以为小反刍兽疫防控与净化提供一定的帮助。目前实验室诊断PPRV的方法主要有病毒的分离鉴定、中和试验、琼脂凝胶免疫扩散试验、ELISA、免疫荧光抗体试验、RT-PCR等方法[11],这些检测方法在病原检测方面都存在一定的缺点,如病原分离耗时长,中和试验特异性低,琼脂凝胶免疫扩散试验、ELISA等方法尚无法鉴别牛瘟病毒和小反刍兽疫病毒。随着分子生物学技术的发展,用荧光标记方法检测特异性产物的实时荧光定量PCR技术,因其灵敏度高、重复性好、定量准确、耗时短、过程封闭、操作简单、成本相对较低等众多优点,逐渐在病原体检测、目的基因表达量、疫苗质量控制等方面得到广泛的使用。
在实时荧光定量PCR试验过程中,要注意做好防污染工作,尽管试验操作比较简单,但操作中的污染如气溶胶的污染会出现假阳性的结果;在制作标准模板时,要使阳性质粒完全混合均匀,以此保证标准模板的浓度梯度正确性。由于荧光染料能够被嵌入到任何脱氧核糖核酸的双链间,所以RT-qPCR荧光染料法的引物在设计时要认真考虑长度、退火温度、保守性、二聚体、发夹结构等方面的问题,尤其是其中的引物二聚体和发夹结构问题,其关系到定量的准确性。以上方面考虑的越周全,得到的结果越稳定,越可靠[12]。Eva Green作为饱和荧光染料,没有“染料重排”的缺点,熔解曲线的分辨率相对较高,而且可以在扩增过程当中及时的从DNA双链中释放出来,从而降低了对PCR扩增过程的影响,因此使用Eva Green染料定量,比使用SYBR染料准确性高。故此,本研究选取Eva Green作为RT-qPCR的荧光染料。
本研究根据NCBI上登录的PPRV的N基因,设计了一对扩增的目的片段为244 bp的特异性引物,构建阳性质粒标准模板,使用Eva Green作为信号报告荧光,建立PPRV RT-qPCR的检测方法。结果表明,该检测方法的标准曲线线性关系良好,熔解曲线峰型单一;特异性强,只能检测出PPRV,最低检测拷贝数为2.46×101copies/μL;重复试验变异系数在1%之下,重复性良好;用建立的RT-qPCR方法检测16份PPRV的阳性样本,结果均为阳性;整个RT-qPCR扩增过程只需45 min,时间过程短。综上所述,本研究建立的PPRV RT-qPCR检测方法,具有良好的特异性、灵敏性和重复性,而且耗时短,可用于临床检测小反刍兽疫病毒感染,对小反刍兽疫的防控提供一定的技术支持。
