副产氨水中(三氮唑合成)甲酰胺含量测定方法研究
2018-12-05杨志刚吴中明刘志勇
杨志刚,吴中明,刘志勇
(滨海县环境保护局,江苏 滨海 224500)
甲酰胺(分子式:HCONH2),外观为无色透明油状液体。它是化工合成中一个重要原料或中间体,在三唑系列的农药、医药生产中有着极其广泛的用途和独特作用[1],也是三氮唑合成中的重要生产原料[2],由于生产中引入甲酰胺,毒性较大,在其副产氨水中要加以控制,采用液相色谱法测定尚未见报道[3]。江苏剑牌农化分析研究中心通过大量的探索、研究,发现当加大流动相中的水相(0.01 mol/L磷酸二氢钾水溶液)的比例,在195 nm波长下可以准确对甲酰胺的含量进行检测,其他相关杂质同时也能得到很好的分离,分析结果能满足定量分析方法验证要求,适用于三氮唑合成生产过程中副产氨水中限量杂质甲酰胺的质量控制检测,反应过程如下:

1 实验操作部分
1.1 所用仪器与试剂
Agilent 1260液相色谱仪,配有二极管阵列检测器和自动进样器;Agilent 色谱工作站;METTLER XSE205电子天平;乙腈(色谱纯);水为新蒸二次蒸馏水;甲酰胺标样(99.0%),江苏剑牌实验室提纯;三氮唑标样(99.0%),江苏剑牌实验室提纯;三氮唑合成副产氨水试样,江苏剑牌农化股份有限公司提供。
1.2 色谱条件
色谱柱:Agilent ZORBAX SB-Aq 250 mm×4.6 mm,5 μm不锈钢柱;流动相:乙腈+0.01 mol/L磷酸二氢钾水溶液(体积比2∶98);流速0.5 mL/min;检测波长195 nm;进样体积:5.0 μL;柱温:30 ℃;保留时间约6.6 min。
1.3 测定步骤
1.3.1 标样溶液的配制
准确称取甲酰胺标样0.02 g(精确至0.000 2 g)到100 mL量瓶中,加水至刻度,摇匀。移取1 mL到50 mL容量瓶中,加水定容到刻度线,混合均匀后备用。
1.3.2 试样溶液的配制
用以上同一天平准确称取副产氨水试样0.2 g(精确至0.000 2 g)到50 mL容量瓶中,用水定容,充分混合后过滤,把滤液作为试样溶液。
1.3.3 进样测定
待仪器稳定后,相邻两针标准溶液中色谱峰面积相对变化小于1.0%时,进样分析。注意按照色谱分析进样要求,进试样前后均要进标准溶液。甲酰胺标准溶液以及副产氨水试样溶液典型谱图如图1~2所示。
1.3.4 含量计算
副产氨水试样中甲酰胺的质量分数X(%)按下列公式计算:
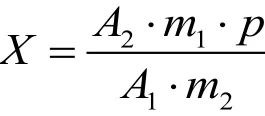

图1 甲酰胺标准溶液色谱图
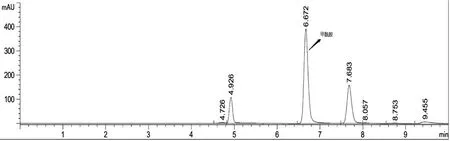
图2 副产氨水试样色谱图
公式中:A1—两针标准溶液中,测得甲酰胺峰面积的平均值。
A2—两针试样溶液中,测得甲酰胺峰面积的平均值。
m1—称取甲酰胺标样的质量,g。
m2—称取副产氨水试样的质量,g。
P—标样甲酰胺的纯度,%。
2 实验的结果与讨论
2.1 检测波长的确定
选用色谱仪Agilent LC-1260(带DAD二极管阵列检测器)进行全波长光谱扫描,如图3所示。当波长为195 nm时,主峰甲酰胺有较强吸收,峰型对称,并且主峰前后无相关杂质峰干扰,所以通过筛选最终确定了195 nm为本方法测定的最佳检测波长。
2.2 流动相条件的选择
为保证三氮唑等杂质峰与主峰甲酰胺完全分离,本实验分别选取了不同比例的甲醇+缓冲盐水溶液、乙腈+缓冲盐水溶液等作为其流动相,发现用乙腈+ 0.01 mol/L磷酸二氢钾水溶液,各组分的色谱峰峰形对称尖锐,有较好的分离度,且基线噪声小,保留时间适宜,最终确定乙腈+0.01 mol/L磷酸二氢钾水溶液(体积比2∶98)作为本试验方法的最佳流动相。
2.3 分析方法的线性关系
分别移取5组不同体积的甲酰胺标样溶液,配成系列对照品溶液,进样5.0 μL(见表1)。用峰面积(y)为纵坐标,质量浓度(x,mg/L)为横坐标绘制线性曲线(见图4),线性方程为y=29.24x+4.791,相关系数为r=1.00,线性范围为0.56~56.6 mg/L。该条件下最低检测限为0.1 mg/L。
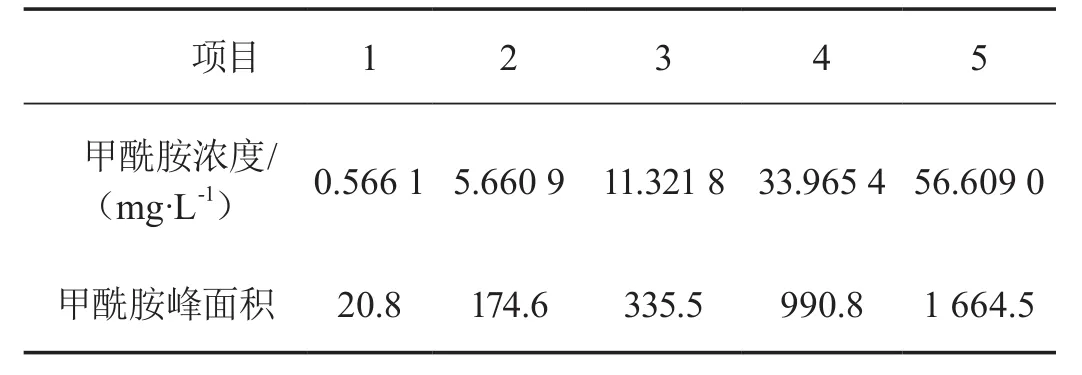
表1 线性关系实验结果

图3 甲酰胺紫外吸收光谱图

图4 甲酰胺线性关系
2.4 分析方法的精密度
随机抽取了一批该产品的代表性试样,分别称量5个样,采用以上选定的色谱条件进行色谱检测,结果如表2所示,该方法的精密度很好,甲酰胺标准偏差为0.003 1,变异系数为2.352%。
2.5 方法准确度试验
在已知含量的典型试样中分别加入5组不同量的甲酰胺标样,进行加标回收率测定。结果甲酰胺的平均回收率可以达到99.59 %(见表3)。

表2 甲酰胺精密度实验结果
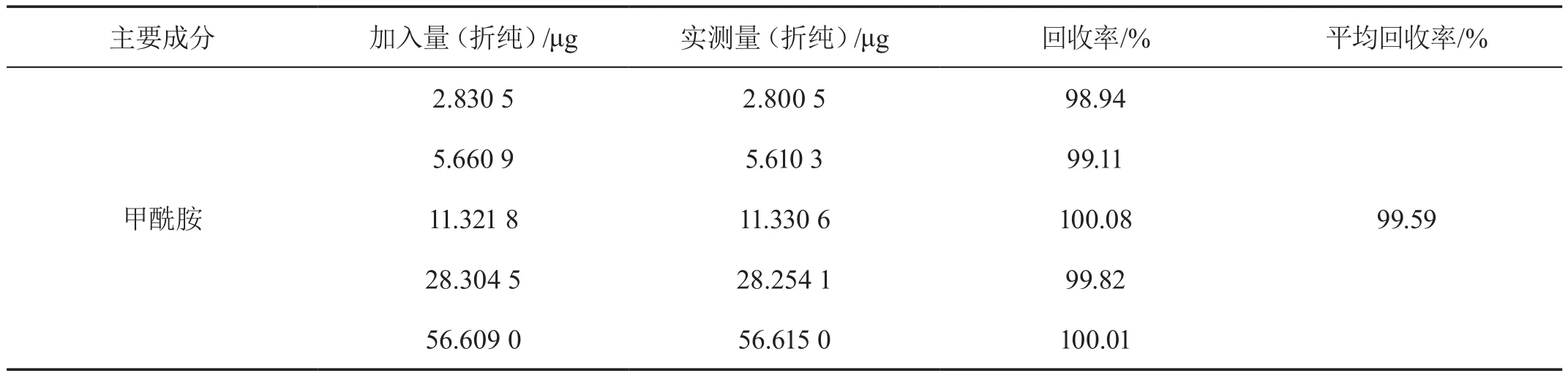
表3 甲酰胺准确度实验结果
