葡萄DFR蛋白的生物信息学分析及自激活检测
2018-12-04刘海霞温灏宇杨波李晓慧张鹏飞温鹏飞
刘海霞,温灏宇,杨波,李晓慧,张鹏飞,3,温鹏飞,3*
(1.山西农业大学 园艺学院,山西 太谷 030801;2.西北农林科技大学 动物科技学院,陕西 杨凌 712100;3.果树种质创制和利用山西省重点实验室,山西 太原 030031)
原花色素(PAs),以黄烷-3-醇的结构为基础,是一类以低分子量单体或高聚合度的聚合体形式存在的无色酚类化合物[1],是类黄酮生物合成途径一个分支的末端产物,对葡萄果实和葡萄酒的颜色、风味、收敛性以及苦味的形成起着重要的作用。前人研究表明,PAs合成涉及类黄酮生物合成途径的一系列酶促反应[2],其中二氢黄酮醇-4-还原酶(DFR)是原花色素合成的关键酶之一,以二氢栎皮黄酮(DHQ)、二氢堪非醇(DHK)和二氢杨梅黄酮(DHM)为底物催化生成原花色素单体黄烷-3-醇,后经聚合而形成原花色素。MYB转录因子参与植物的次生代谢(如黄酮类物质的合成[3])、细胞分化、细胞周期、器官形态建成以及激素和逆境因子应答等过程[4]。目前,已经证实MYB转录因子(如MybPA1)通过控制原花色素生物合成过程中相关结构基因的时空表达,从而调控原花色素的生物合成[5,6]。但MybPA1与原花色素合成关键酶DFR的关系尚未见报道,酵母双杂交是用来研究蛋白与蛋白之间相互作用的有效方法,构建诱饵质粒是酵母双杂交的基础[7]。为了进一步明确DFR蛋白的作用和功能,本研究对DFR的氨基酸序列进行了生物信息学分析,并构建了DFR基因的酵母双杂交诱饵载体,同时检验其DFR蛋白的自激活活性,以期通过酵母双杂交技术来研究与DFR有互作反应的蛋白,从而为原花青素等酚类物质的生物合成及调控机制研究奠定重要基础。
1 材料与方法
1.1 试验材料
酵母菌AH109、表达载体pGBKT7购于Clontech公司,限制性内切酶EcoRI、SacI、T4-DNA连接酶、DNA marker、克隆载体pMD19-T购于TaKaRa公司。提取质粒和回收胶试剂盒购于Omega公司,大肠杆菌感受态细胞DH5α、高保真酶定购于全式金生物技术公司。所有的测序及引物合成都来源于华大有限公司。
1.2 葡萄DFR蛋白生物信息学分析
利用在线软件InterProScan程序检索EBI的InterPro数据库(http://www.ebi.ac.uk/interpro/)预测DFR蛋白序列上潜在的结构域和功能位点。利用NCBI(https://www.ncbi.nlm.nih.gov/)上保守区域CDD数据库分析了DFR蛋白的保守结构域(http://www.ncbi.nlm.nih.gov/cdd/wrpsb.cgi)。在线软件STRING(http://string-db.org/cgi/input.pl)对DFR蛋白互作进行预测。
1.3 DFR基因片段引物设计及PCR扩增
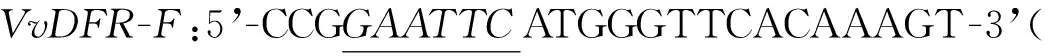
1.4 诱饵载体pGBKT7-DFR的构建与检测
将DFR扩增回收产物在16 ℃下与克隆载体pMD19-T进行连接反应45 min,然后在冰浴条件下转化到感受态细胞DH5α中,挑取阳性菌落进行PCR验证,成功获得克隆载体pMD-DFR。提取pMD-DFR质粒,用内切酶SacI和EcoRI双酶切pMD-DFR和空载体pGBKT7质粒,割胶回收所切出的目的片段,然后在16 ℃下与T4-DNA连接酶连接反应10 h,再迅速转化到感受态细胞DH5α中,筛选出阳性菌珠后进行菌液PCR验证,将PCR验证后的菌液过夜揺菌扩大培养后提取质粒进行双酶切鉴定。
1.5 诱饵质粒pGBKT7-DFR的自激活活性检测
将1.2 μg诱饵质粒pGBKT7-DFR中加入10 μL pGADT7空质粒和10 μL预变性的鲑鱼精DNA(沸水浴5 min,冰浴2 min,重复3 次)在预冷的1.5 mL离心管中混匀,加入200 μL的酵母菌感受态细胞AH109后迅速进行涡旋震荡。最后再加入650 μL过夜灭菌后的PEG/LiAc立刻漩涡震荡25 s,置于摇床中在30 ℃,220 r·min-1下培养1 h。然后加入80 μLDMSO后缓慢上下颠倒混匀,42 ℃下水浴12 min后迅速冰浴4 min,在室温条件下,10 000 r·min-1,离心45 s后将上清液迅速倒掉,向沉淀中加入300 μL灭菌后的双蒸水进行涡旋混匀,共转化于感受态细胞AH109中,作为试验组。同时以共转化pGBKT7与pGADT7空质粒作为空载体对照,共转化pGBKT7-lam与pGADT7-T作为阴性对照,共转化pGBKT7-53与pGADT7-T作为阳性对照。30 ℃恒温下,分别将空载体组、阴性对照组、阳性对照组、试验组的转化液涂在缺陷型SD/-Trp/-Leu、SD/-Trp/-Leu/-His/-Ade板上和含有X-α-Gal的SD/-Trp/-Leu和SD/-Trp/-Leu/-His/-Ade培养基上培养2~3 d,然后观察菌落的颜色变化。
1.6 pGBKT7-DFR诱饵载体的毒性检测
将转化酵母AH109的pGBKT7和pGBKT7-DFR阳性克隆菌液分别涂布于SD/-Trp营养选择性固体培养基中,30 ℃倒置培养2~4 d,待长出菌落后,分别随机挑取5 个单菌落于SD/-Trp营养选择性液体培养基中,30 ℃,220 r·min-1恒温培养24 h,测培养物的OD600值,如果OD600≥0.8,则诱饵载体无毒性,如果OD600<0.8,则诱饵载体对酵母菌有毒性。
2 结果与分析
2.1 DFR蛋白功能域的分析
通过在线InterProScan软件中检索的EBI数据库分析,结果表明,DFR蛋白具有NAD依赖型的差向异构酶、脱氢酶N端结构区域(NAD-dependent epimerase/dehydratase,N-terminal domain)和NAD(P)结合位点(图1)。利用NCBI中Blast在线检索的CDD数据库,发现DFR蛋白具有NADB-Rossmann保守结构域,属于黄酮还原酶家族成员,拥有NADP、活性和底物的结合位点和独特的AR-like-SDR-e元件,具备了还原酶的特征结构序列(图2)。

图1 DFR功能位点分析Fig.1 Functional sites of DFR

图2 DFR蛋白保守结构域分析Fig.2 Analysis of conserved domains of DFR protein
2.2 DFR蛋白互作预测
利用STRING在线软件对DFR蛋白与MybPA1蛋白互作情况进行预测分析,图3中的互作网络显示DFR蛋白与MybPA1蛋白存在互作关系,同时DFR与原花色素生物合成相关酶基因(F3’5’H、ANR、LODX、F3H、LAR)也存在互作关系。

图3 DFR蛋白互作网络预测Fig.3 DFR protein interaction network prediction
2.3 DFR基因PCR扩增
菌液PCR扩增后,用1%的琼脂糖凝胶电泳跑胶检测可发现1 条大小约为1 014 bp片段(图4),和预期结果相同。
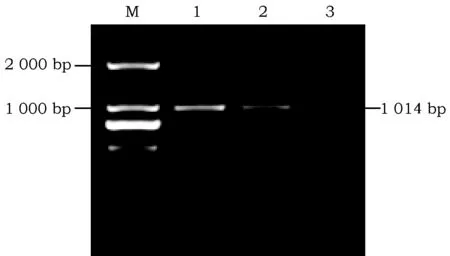
图4 DFR基因片段PCR产物Fig.4 PCR product of DFR gene fragmentM:2000 DNA marker;1~3:菌液PCR产物M:2000 DNA marker;1-3:PCR product of bacterial solution
2.4 pMD-DFR克隆载体的构建及鉴定
将切胶回收后的DFR产物在16 ℃下与克隆载体pMD19-T反应45 min,快速转化到大肠杆菌感受态细胞DH5α中,在37 ℃过夜培养待长出大量菌落时,挑取饱满的单菌落进行菌液PCR阳性筛选与鉴定,可见到与预期相一致的目的片段(图5),测序验证为目的基因,且无碱基突变,成功构建了克隆载体pMD-DFR。

图5 菌液PCR鉴定Fig.5 Identification of bacteria by PCRM:2000 DNA maker;1~4:菌液PCR产物M:2000 DNA Marker;1-4:PCR productof bacterial solution
2.5 pGBKT7-DFR诱饵表达载体的构建及鉴定
将空质粒pGBKT7和成功构建的克隆载体pMD-DFR用EcoRI和SacI内切酶进行双酶切回收目的片段并用T4-DNA连接酶连接后,转化大肠杆菌DH5α,得到单克隆转化子菌株1和2,采用PCR与双酶切均检测到质粒中插入一段与目的基因大小相当的片段(图6),表明诱饵载体pGBKT7-DFR构建成功。
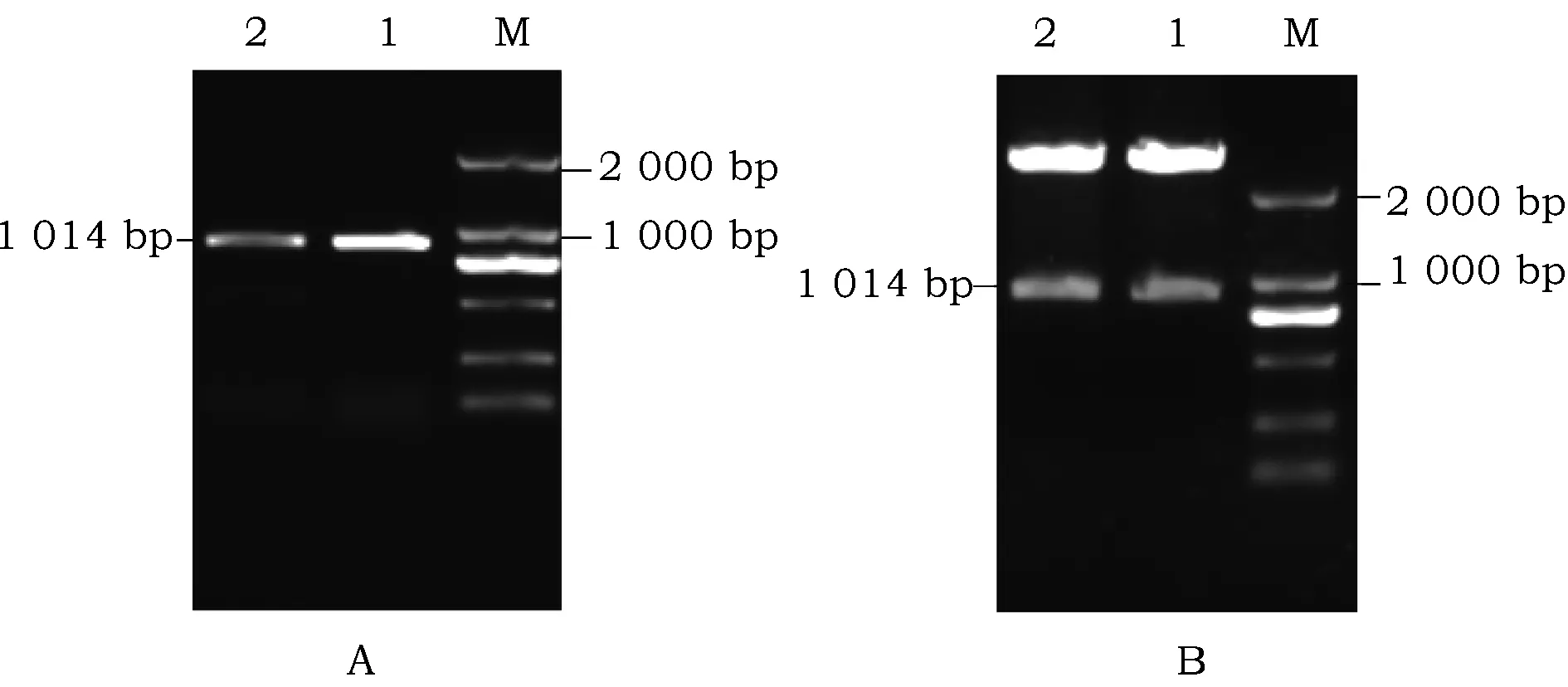
图6 pGBKT7-DFR诱饵载体PCR和双酶切鉴定Fig.6 Detection of bait vector pGBKT7-DFR with PCR and double enzyme digestion注:A:pGBKT7-DFR诱饵质粒PCR验证;M:2000 DNA ladder;1~2:PCR扩增产物;B:pGBKT7-DFR诱饵质粒的双酶切鉴定;M:2000 DNA ladder;1和2:双酶切结果Note:A:Bait vector pGBKT7-DFR PCR identification;M:2000 DNA ladder;1-2: PCR amplification products;B:Detection of bait vector pGBKT7-DFR by doub le enzyme;M:2000 DNA ladder;1-2:results of double enzyme
2.6 pGBKT7-DFR诱饵载体的毒性和自激活检测
分别挑取转化质粒pGBKT7和pGBKT7-DFR的酵母菌AH109于SD/-Trp液体培养基中,30 ℃,220 rpm下振荡培养24 h,用紫外分光光度计测定在波长为600时培养物的OD值。由表1可见,两者菌液的OD600值都大于0.8,说明诱饵载体pGBKT7-DFR对AH109没有毒性作用。分别吸取160 μL空载体对照组、阴性对照组、阳性对照组、试验组的菌液于含有显色剂X-α-Gal的SD/-Trp/-Leu与SD/-Trp/-Leu/-His/-Ade板上,放置于30 ℃的恒温培养箱中,2 d后发现只有阳性对照组长出蓝色菌落,而其他组在SD/-Trp/-Leu/X-α-Gal板上长出白色的菌落,在SD/-Trp/-Leu/-His/-Ade/X-α-Gal板上无菌落生长(如图7),表明诱饵质粒pGBKT7-DFR无法激活AH109酵母菌中指示基因MEL1的表达(MELI编码α-半乳糖苷酶,随着显色剂X-α-Gal的出现阳性菌落就会由白色变成蓝色),因此证实了pGBKT7-DFR诱饵蛋白没有自激活活性。
3 讨论与结论
二氢黄酮醇-4-还原酶(DFR)拥有特定的NADPH结合域[8]。DFR基因编码的蛋白具有NAD(P)结合位点,在NADPH存在的环境中,DFR可以特异性的把二氢黄酮醇催化成黄烷-3,4-二醇(即隐色花色素),是类黄酮代谢中原花色素合成的关键酶之一。自1985年首次从玉米和金鱼草中分离以来,DFR基因先后从矮牵牛、拟南芥[9]、油菜、水稻、马铃薯、非洲菊、玫瑰、玉米[10,11]等草本植物,以及甜樱桃[12]、石榴、山葡萄、猕猴桃[13]、山竹子、荔枝[14]等果树中得到了克隆。

表1 诱饵质粒的OD600值Table 1 The OD600 value of bait plasmid
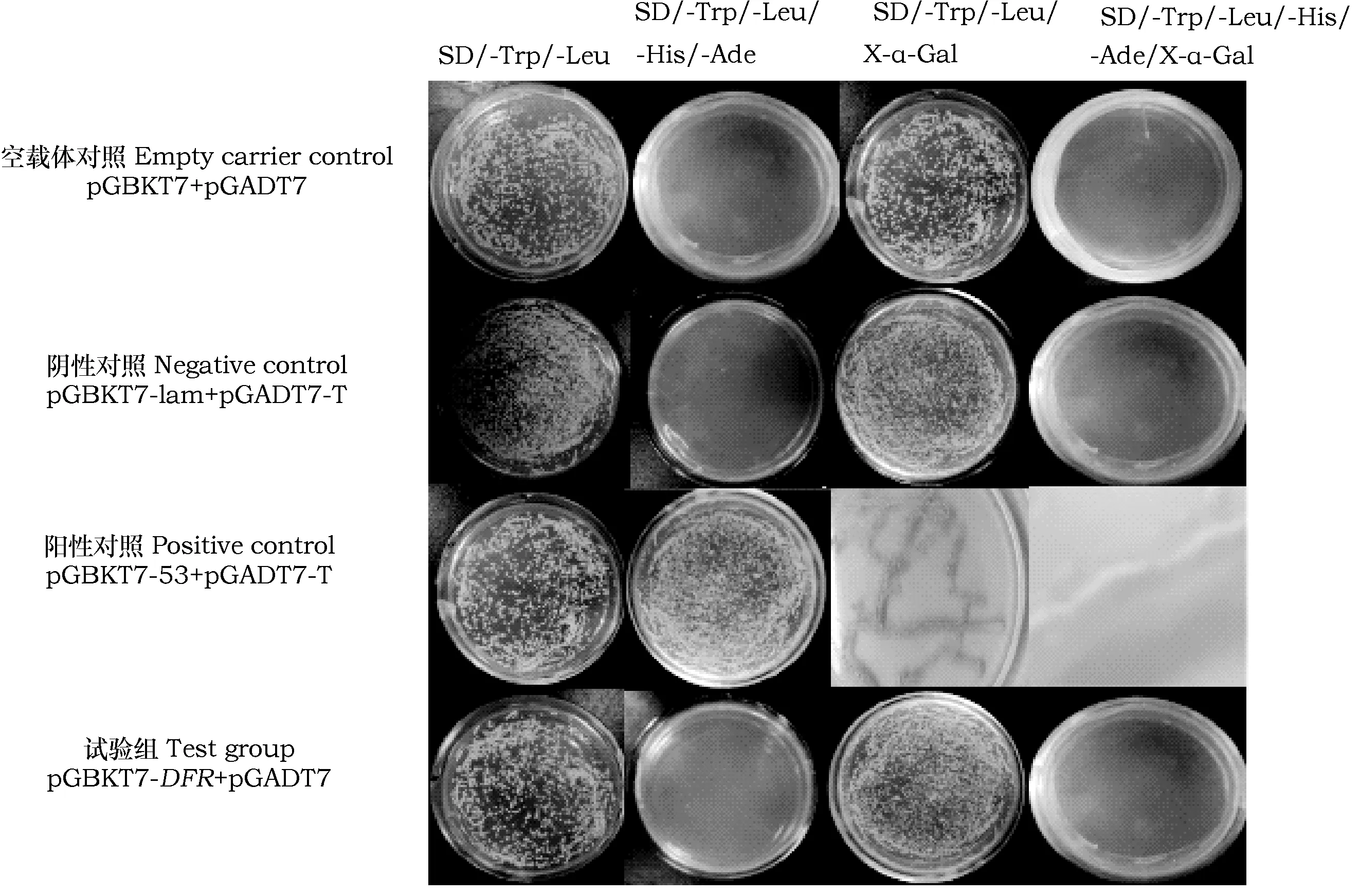
图7 重组质粒转化酵母AH109细胞自激活活性检测结果Fig.7 Results of self-activation activity of yeast AH109 cells transformed by recombinant plasmid
DFR基因广泛分布于各植物的不同组织中,有研究发现该基因表达于枸杞不同的组织中,如根、茎、叶等。除了影响着原花色素的生物合成,还在单宁、花色苷等酚类物质的合成途径中起着重要作用[15]。潘丽晶等[16]发现:石斛兰的花中明显缺少蓝色和黄色,这种现象与DFR的活性有着不可分割的关联。有研究证实,在赤霞珠葡萄果实中,DFR的活性和基因表达水平直接影响着葡萄果实原花色素的生物合成[17],在原花色素合成途径中起着至关重要的作用[18,19]。本课题组前期研究结果表明:DFR和MybPA1在葡萄的不同时期都有表达,MybPA1及其MybPA1蛋白的表达与DFR基因呈负相关[20]。王晓平等人[21]的研究表明,京亚葡萄中的MybPA1、MybPA2与原花色素的形成有关,Bogs等人[6]研究MybPA1对西拉葡萄中的原花色素的生物合成调节有特效。由于MYB 转录因子往往是通过调控结构基因表达而影响原花色素合成[22],所以推测MybPA1可能是通过调节DFR的表达从而调控原花色素的合成。同时从在线软件STRING网络预测图中得出,MybPA1蛋白与DFR蛋白存在互作关系,但是互作机制还未明确需要更深一步的研究。
酵母双杂交技术是研究蛋白与蛋白互作的一种有效方式,此技术不仅用于研究已知蛋白间的相互作用,在筛选与诱饵蛋白互作的未知蛋白方面也起着关键作用[23]。构建诱饵表达载体是利用酵母双杂交系统大量筛选互作蛋白的基础。本研究通过生物信息学分析得出,DFR蛋白属于黄酮还原酶家族成员,拥有NADB-Rossmann保守结构域和NADP结合位点,具有还原酶的典型特征;从预测蛋白互作网络图中得出,DFR蛋白与MybPA1蛋白存在互作关系。成功构建了酵母双杂交诱饵载体pGBKT7-DFR,细胞生长分析显示DFR对酵母菌AH109无细胞毒性,通过检测AH109中报告基因MEL1的活性,证明诱饵载体pGBKT7-DFR对酵母菌AH109无自激活,该诱饵载体可用于筛选酵母双杂交系统中与DFR互作的蛋白,从而为深入研究DFR的功能奠定了重要基础。
