脊髓小脑性共济失调一家系报道
2018-11-15重庆医科大学附属南川人民医院神经内科重庆408400
陈 莉(重庆医科大学附属南川人民医院神经内科,重庆408400)
脊髓小脑性共济失调(SCA)是遗传性共济失调的主要类型,是一种起病隐匿、逐步进展、高度遗传异质性神经系统变性病,主要累及人类中枢神经系统。成年起病、呈常染色体显性遗传及小脑性共济失调为本病的共同特征。现报道本病有30余种亚型,我国最常见为SCA3型。现报道如下。
1 临床资料
1.1 病例介绍 患者,女,40岁,主因“步态不稳、吐字不清3年”于2017年3月16日收入本院神经内科。主要表现:患者3年前无明显诱因出现走路摇晃,步态不稳,伴头晕、视物模糊,行走时向前冲,向后跌倒1次,伴双上肢精细动作缓慢,讲话缓慢,吐字不清,饮水呛咳,无头痛、呕吐,无肢体抖动、身体发僵。2年前出现睡眠中大喊大叫,肢体舞动,噩梦,清醒时无幻觉。半年前出现便秘,伴记忆力明显减退。病程中无情绪低落、肢体疼痛。既往体健,否认脑卒中、创伤史。家族史:患者的外婆、姨姥姥、舅舅、母亲、二姨、舅舅的2个女儿、二姨的2个儿女均有类似症状,患者的儿子智力较同龄人稍差。
查体:右侧卧位血压 122∕84 mm Hg(1 mm Hg=0.133 kPa),心率 72 次∕分,立位血压 115∕76 mm Hg,心率79次∕分,内科系统查体无阳性体征。神经系统查体:神志清楚,构音障碍,计算力、记忆力减退,反应迟钝,颅神经查体无阳性体征。四肢肌容积正常,四肢肌力5级,四肢肌张力减低,双侧指鼻试验、跟膝胫试验欠稳准,轮替试验动作幅度及速度减慢,闭目难立征阳性。行走时步基增宽,醉酒样步态,后拉试验阴性。双侧针刺觉及音叉振动觉对称。四肢腱反射未引出。双侧Hoffmann征、Rosolimo征、掌颏反射、Babinskin征均阴性。辅助检查:头颅CT可见小脑萎缩(图1)。三大常规、凝血分析、肝肾功能、电解质、血脂、血糖、叶酸、维生素B12、铁蛋白、肿瘤标志物、抗O、类风湿因子、C反应蛋白、血细胞沉降率(血沉)、乙肝五项、糖化血红蛋白、甲功全套、蛋白电泳、免疫全套化验均正常。血清同型半胱氨酸 18.1 μmol∕L。胸部 X 线片、心电图、腹部彩色多普勒超声(彩超)、妇科超声、泌尿系超声、经胸心脏超声心动图均正常。黑质回声强度Ⅱ级。眼科会诊未见KF环,眼底照相正常。进一步完善头颅磁共振成像(MRI)检查仍可见小脑萎缩改变(图2)。颈椎MRI检查脊髓未见异常信号改变(图3)。患者均成年起病,临床以共济失调症状体征为主,影像学检查可见小脑萎缩,结合家系调查代代均发病,男女均患病,且发病年龄逐渐提前,具有遗传早现表现,故临床诊断为共济失调综合征:SCA可能性大。患者由于经济原因未进一步行基因检测,无法明确具体分型。告知患者及家属病情后放弃治疗回家休养,患者出院时症状无明显缓解。3个月后随访,患者病情无明显变化,6个月后随访,患者步态不稳加重,需在家人搀扶下缓慢行走,跌倒2次。目前仍在随访中。
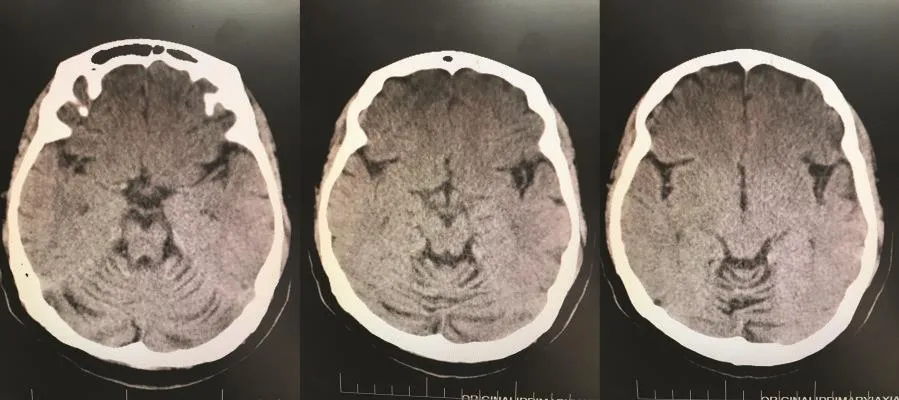
图1 SCA先证者头颅CT检查示小脑蚓部和双侧小脑半球明显萎缩
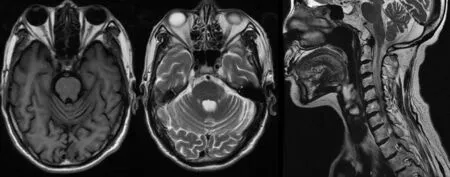
图2 SCA先证者头颅MRI检查示小脑蚓部和双侧小脑半球萎缩
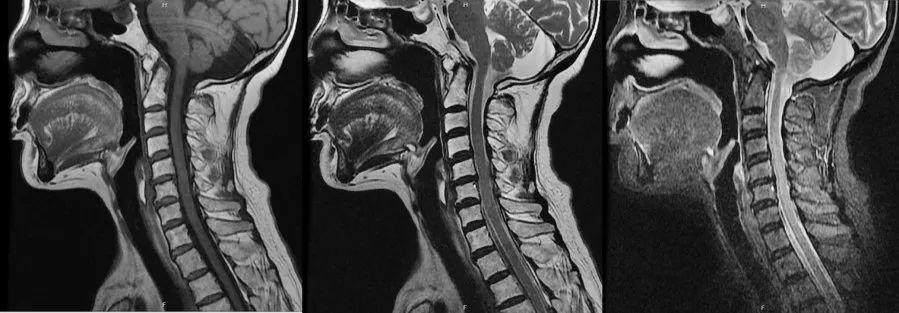
图3 SCA先证者颈椎MRI检查示颈髓未见异常信号改变
1.2 家系调查 该患者家系4代共29人,均无近亲结婚,患病10例(男2例,女8例)。先证者的外婆(Ⅰ2,55岁发病,已故)、姨姥姥(Ⅰ1,50岁发病,已故)、舅舅(Ⅱ2,44岁发病,已故)、母亲(Ⅱ5,54岁发病,已故)、二姨(Ⅱ3,48岁发病,已故)、舅舅的两位女儿(Ⅲ1、Ⅲ2,分别于 42、39岁发病)、二姨的两位儿女(Ⅲ6、Ⅲ7,分别于39、38岁发病)均有类似症状,患者的儿子智力较同龄人稍差,未发病。第Ⅰ代平均发病年龄52.5岁,第Ⅱ代48.67岁,第Ⅲ代39.6岁。第Ⅰ代和第Ⅱ代患者全部去世,第Ⅳ代目前未发病。SCA患者家系图谱见图4。
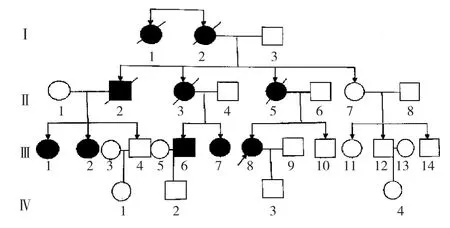
图4 SCA患者家系图谱
2 讨 论
SCA是一大类由于遗传因素造成的神经系统变性疾病,多为常染色体显性遗传,占神经系统遗传性疾病的 10%~15%,患病率为(1∕10~4∕10)万,各种族均可见[1]。共同临床特征表现为成年起病、呈常染色体显性遗传及小脑性共济失调等,具有在连续数代中发病年龄提前和病情加重的特征(遗传早现现象)[2]。SCA有明显的遗传异质性和基因多效性,使患者临床表现错综复杂[3]。目前,发现SCA1~29等若干亚型均为单基因遗传病,已经发现30多种致病基因位点[4],我国常见的SCA亚型为SCA3,基因突变位点在14q24.3~q32,以小脑性共济失调、锥体系和椎体外系症状、进行性眼外肌麻痹、远端肌萎缩等为临床特征[5]。
目前,认为SCA的发病机制可能与多聚谷氨酰胺扩增、非编码区扩增、常规突变有关。由于SCA不同亚型基因突变位点的不同导致该病不同亚型损伤部位和发病机制不同,临床表现也各异,在基因突变和临床症状上既有重叠又有差异,称之为SCA的多态性和异质性[6]。基因检测是确定SCA基因亚型的唯一方法。
SCA以小脑、脑干和脊髓变性萎缩为共同的病理改变。大体临床表现为:30~40岁隐匿起病,缓慢进展;首发症状多以双下肢共济失调为主,表现为走路摇晃、步基增宽或突然跌倒,伴有双手笨拙及意向性震颤、辨距不良、构音障碍、眼球震颤等。查体可见肌张力障碍、锥体束征和深感觉障碍[7]。该病例家系中无近期结婚现象,男女均有发病,发病年龄38~55岁,以相似症状起病,后代较前代发病年龄逐渐减小,符合遗传早现现象。SCA患者的CT或MRI可提示小脑和脑干萎缩。但SCA各亚型临床表现大多相互重叠,发病初期影像学检查可无异常,仅根据临床表现和影像学检查无法进行精确分型。基因检测为确诊SCA的“金标准”,但对于基层医院大多数以农村患者为主,无条件完善基因检测时仍以临床诊断为主,需临床医生对该疾病提高认识。
目前,SCA尚无完全治愈的方法,临床上以对症药物治疗为主,目前研究较为热门的治疗方法为干细胞移植[8-9],但临床效果欠佳,还有待进一步研究。SCA和其他神经系统变性病一致,其主要治疗目的为减轻患者临床症状、延缓疾病恶化速度、提高患者的生活质量[10]。
综上所述,对于以共济失调症状为首发症状就诊的患者,临床医生必须详细询问病史,仔细对患者进行体格检查,尤其是对于存在神经系统症状合并家族史的患者,应考虑到神经系统遗传病的可能。在排除其他导致共济失调的病因后,进行基因检测有助于明确诊断[11]。同时,对SCA家族史中的健康人及早进行基因检测,有助于预防和延缓疾病的发生、发展[12]。SCA患者及家族中健康人在结婚生育时鼓励进行遗传咨询和产前筛查,尽可能地减少患儿出生[13],是优生优育、预防缺陷儿出生的重要手段。
