校正肿瘤纯度影响的差异表达基因分析
2018-11-12张伟伟吴志强
张伟伟, 吴志强
(东华理工大学 理学院,江西 南昌 330013)
癌症是一类由于细胞分裂和凋亡机制失常而导致的疾病,通常表现为恶性肿瘤(Hanahan et al., 2011)。差异表达的基因可能直接或间接关系到癌症的发生和易感性。差异表达基因的检测能够从基因层次揭示癌症的发生机理并且能够更好地探索癌症治疗的新方法(Wu et al., 2013)。
肿瘤样本的差异表达分析通常是将肿瘤样本和正常样本直接进行比对。然而,临床上获得的肿瘤组织往往包含不同比例的正常细胞,如果不考虑正常细胞的混入,直接进行差异甲基化/基因表达或样本聚类等下游分析,势必会导致结果出现偏差甚至错误。在表观遗传层面上,针对肿瘤纯度导致差异甲基化分析出现偏差的问题,Zheng等(2017)提出了肿瘤纯度校正的差异甲基化分析模型 InfiniumDMC。Zhang等(2017)提出了一种考虑肿瘤细胞纯度的肿瘤样本亚型聚类方法 InfiniumClust。同样,肿瘤样本的“不纯”也会使肿瘤样本在基因上的均值估计产生偏差,进而导致差异表达分析结果出现偏差。为此Aran等(2015)将肿瘤纯度作为线性“协变量”加入回归模型,尽管这样做得到了更多差异表达的基因。但是,通过分析发现,肿瘤纯度与基因差异之间的关系是乘性的而非原来认为的线性关系。忽视肿瘤纯度,或者将肿瘤纯度作为“协变量”加入回归模型都会使差异分析结果出现偏差。
为解决上述问题,本文在正态分布假设下构建了肿瘤样本基因表达量的混合概率模型,通过广义最小二乘法和Wald检验来确定每个基因在癌症和正常细胞之间的差异性,并通过TCGA数据分析来说明所提模型的优越性。
1 考虑肿瘤细胞纯度的差异表达基因模型
基因的原始数据不服从正态分布,经过log2变换后数据的正态性会变强。图1以肺腺癌为例,图1a显示肺腺癌正常样本变换前后基因正态分布拟合检验的统计量,每个点代表一个基因,显然变换后的统计量变得更加小,特别是对于那么违反正态假设的基因,因此log2变换有助于数据正态化。其次,对变换后的正常样本进行正态分布拟合检验,图1b显示大部分p值均分分布,这表明正态分布假设对于绝大多数基因是成立的。虽然有一小部分基因具有较小的p值,但这些基因可能是正常样本中不同细胞的差异表达基因。因此,在以下模型假设中可以将每个基因在正常样本中的表达量假设成一个单一的正态分布。
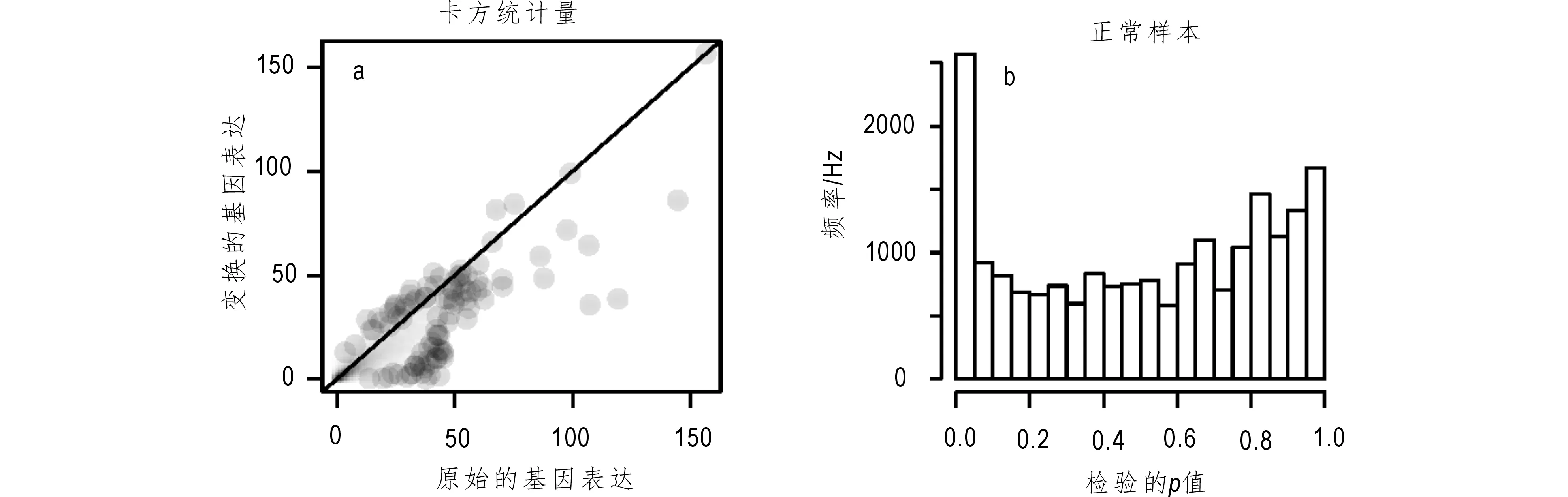
图1 正常样本上所有基因正态分布检验的p值和统计量Fig.1 P-values and statistics for the normal distribution of all genes in normal samples
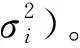
Yis′ =(1-λs)Xis+λsYis
=(1-λs)Xis+λs(Xis+δis)
=Xis+λsδis
令Zi=[Xi1,Xi2,…,Xin0,Yi1′,Yi2′…,Yin1′]。其中前n0项为正常样本的基因表达量,后n1项为肿瘤样本的基因表达量。上述问题变为线性模型Zis=mi+asμi+s,s=1,2,…,n0+n1。模型的参数可以通过广义最小二乘法来求解,令
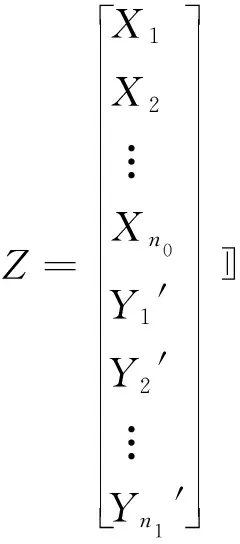
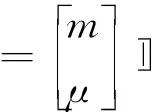
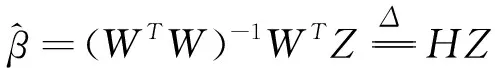
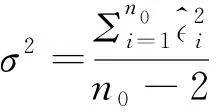
把肿瘤纯度作为线性模型的一个实验设计因子引入,会降低肿瘤样本的内部方差,因此,从理论上来说对于差异表达基因的估计会有所提升。选取 InfiniumPurify估计的肿瘤细胞纯度数据,并将所提方法命名为LinReg,通过与limma (Ritchie et al., 2015)在TCGA的14种癌症类型(正常样本数量超过10个的癌症类型)上的差异分析比较来说明所提方法的优越性。
2 TCGA肿瘤样本差异表达基因分析结果
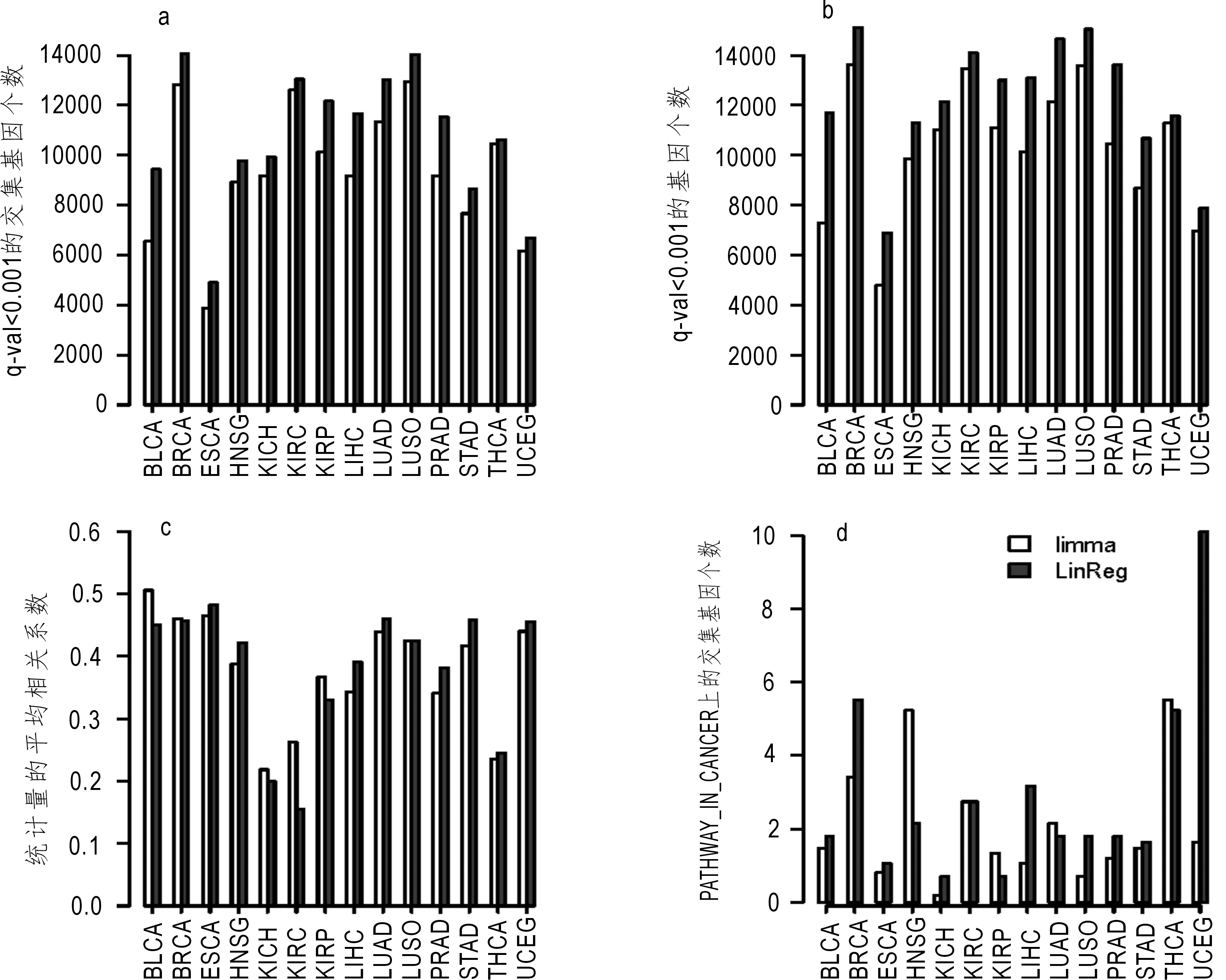
图2 两种方法在不同癌症类型中各指标比对图Fig.2 Comparison diagram of the two methods in different cancer types
首先,把每一种癌症类型的肿瘤样本随机平均分成两份,将这两份肿瘤样本分别和正常样本利用limma和LinReg进行差异表达基因分析(图2)。在同样的显著性指标下(FDR < 0.001),对每一种方法将这两组探测到的差异表达基因取交集,并且来比较一下交集中所包含的基因的数量。图2a显示对于绝大多数TCGA的癌症类型,线性模型比limma得到了更多的交集基因,表明线性方法相比较limma具有更好的探测出诊断生物标志物的性能。接下来比较这两种方法在同样的显著性指标下(FDR < 0.001)得到的差异表达的基因数量。如图2b所示,对于绝大多数TCGA的癌症类型,线性模型比limma得到了更多的差异表达的基因,表明线性方法敏感性更强。虽然不同癌症类型具有不同的病理学基础和差异表达的基因,但从整体上看,不同癌症类型应具有一些相同点,这些共有的性质会带来癌症类型之间较高的统计量相关性。图2c中列举了不同癌症类型与其他癌症类型的平均统计量相关性,结果同样表明线性方法比limma的相关性更强,说明其更具一致性。最后,来分析一下差异表达基因的生物学意义。选取FDR最小的前2 000个基因,将这2 000个基因与KEGG的“PATHWAY_IN_CANCER”上的328个基因取交集,来看一下这14种癌症上两种方法的交集基因个数。图2d显示线性方法在BLCA,BRCA,ESCA,KICH,KIRC,LIHC,LUSC,PRAD,STAD和UCEC这10种癌症上具有更多的交集中的基因,其中LinReg在UCEC上的交集基因的个数几乎是limma的5倍。这些结果说明线性方法得到的基因更具有生物学意义,这些基因对于癌症诊断和治疗具有重要意义。
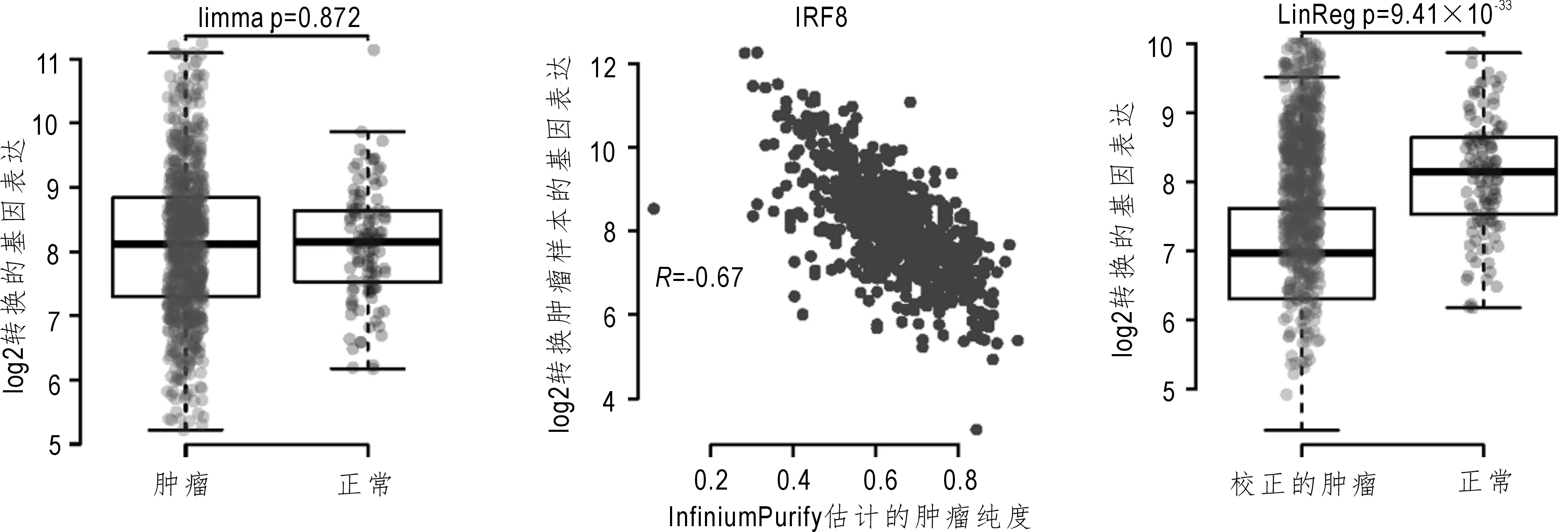
图3 只被线性方法探测到的差异表达的基因与肿瘤纯度的关系Fig.3 The relationship between differentially expressed genes only detected by linear method and tumor purities
为了更好地理解肿瘤纯度在差异表达基因分析中所起的作用,本文任意选取了一个只能被LinReg探测到差异的基因来研究该问题。图3左面面板显示该基因log2变换后在肿瘤样本和正常样本的表达量分布,该基因的均值在两组之间是没有差异的,limma检验的p值为0.872;中间的面板显示该基因在肿瘤样本上log2变换后的表达量和肿瘤样本纯度的相关系数,相关系数达-0.67;右面面板显示校正肿瘤纯度后,两组之间均值有明显差异,利用LinReg进行检验发现p=9.41×10-33。这些结果说明肿瘤样本间较大的组内方差是由于肿瘤样本的纯度引起的,因此利用癌旁的正常样本进行线性提纯后,发现该基因在两组之间具有明显差异,这个结果进一步说明了差异分析时校正肿瘤纯度的必要性。
3 结论
本文针对肿瘤样本“不纯”导致差异分析结果出现偏差的问题,构建了肿瘤样本基因表达量的混合概率模型。该模型是把肿瘤纯度作为影响差异表达基因分析的因素引入差异分析,相比直接使用肿瘤样本作为癌症细胞样本与正常样本直接比对的方法更加合理,有效避免了因为肿瘤样本“不纯”而导致差异表达分析出现的偏差。同样肿瘤纯度也影响着基因共表达网络和共甲基化网络,计划将
在进一步工作中构建肿瘤纯度校正的基因共表达网络和共甲基化网络,并分析其与传统的共表达/共甲基化网络在功能模块分析中的差异。
