5-苯基异噁唑-3-羧酸乙酯的合成研究*
2018-11-06张燕芸齐思佳李洪爽李福荣
张燕芸 齐思佳 倪 雯 李洪爽 李福荣
(泰山医学院药学院,山东 泰安 271016)
异噁唑作为一种普遍存在杂环优势结构骨架,已经表现出广泛的药理学活性,如抗肿瘤[1]、抗菌[2]、抗糖尿病[3]、抗病毒[4]、杀螨[5]等,因此吸引了众多药物化学工作者的广泛关注。传统合成异噁唑杂环的方法主要有:(1) 羟胺类化合物经N-氯代丁二酰亚胺(NCS)氯代后,与炔烃进行[3+2]环加成反应[1];(2) 腈类化合物经氧化后与端基炔烃进行1,3-偶极环加成反应[6]。尽管以上方法为构建功能性异噁唑类化合物做出了较大贡献,但是存在着副产物多、收率较低等缺点,尤其不能直接适用于多酚羟基苯基取代的异噁唑的合成。
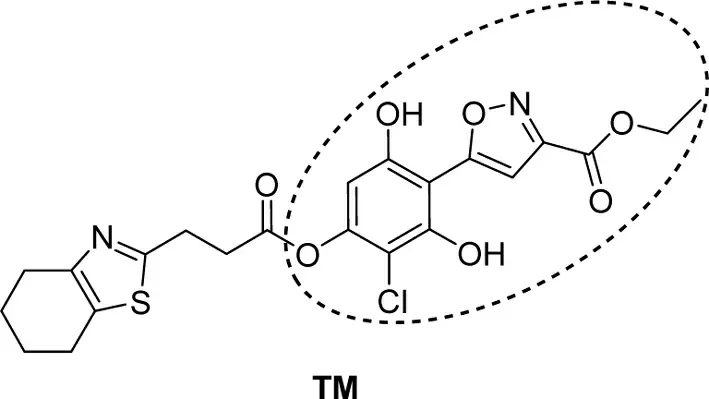
图1 四氢苯并噻唑类小分子TM
笔者课题组一直致力于四氢苯并噻唑类抗肿瘤小分子的合成[7]。为提高此类化合物的抗肿瘤活性,增强其代谢稳定性,笔者设计了四氢苯并噻唑母核缀合异噁唑杂环的化合物(图1,TM),并探索了一条简洁高效的方法合成此化合物的关键中间体。即以1-(3-氯-2,4,6-三羟基苯基)乙酮(A)为起始原料(图2),首先在K2CO3作用下利用溴化苄保护苯环上的三个酚羟基得到中间体(B),随后以NaH作为强碱与草酸二乙酯进行缩合得到烯醇型中间体(C)[8]。C与盐酸羟胺直接缩合即得异噁唑环(D),最后采用BCl3/DCM体系脱除苄基保护基制备得到了目标产物(E)。
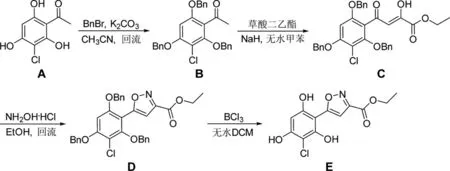
图2 目标产物E的合成路线
1 材料与方法
1.1 材料与试剂
1-(3-氯-2,4,6-三羟基苯基)乙酮,伊诺凯试剂公司产品;溴化苄,百灵威试剂公司产品;草酸二乙酯,阿拉丁试剂公司产品;盐酸羟胺,百灵威试剂公司产品;三氯化硼,阿拉丁试剂公司产品;其他试剂及溶剂均为分析纯,伊诺凯试剂公司产品。柱层析硅胶(200-300目),青岛海洋化工有限公司产品。
1.2 仪器设备
Bruker Avance 400 MHz核磁共振仪(四甲基硅烷为内标,Acetone-d6为溶剂),德国Bruker公司;Agilent 1100 ESI-MS液质联用仪,美国Agilent公司。
2 实验方法
2.1 1-(2,4,6-三苄氧基-3-氯苯基)乙酮(B)的合成
于100 ml圆底烧瓶中,加入1.01 g (5 mmol) 1-(3-氯-2,4,6-三羟基苯基)乙酮(A),溶于60 ml乙腈中,随后加入2.42 g (17.5 mmol)碳酸钾,搅拌5 min后,加入1.9 ml(16.0 mmol)溴化苄。反应液加热回流6 h,冷却至室温,搅拌过夜。反应液过滤,滤饼用50 ml二氯甲烷洗涤,滤液减压浓缩,柱层析纯化,得1.44 g淡黄色固体(B),收率61%。1H NMR (400 MHz,Acetone-d6),δ:7.35~7.54 (m,15H),6.96 (s,1H),5.30 (s,2H),5.23 (s,2H),5.02 (s,2H),2.39 (s,3H);ESI-MS:calculated,472.96;found,473.30,[M+H]+。
2.2 2-羟基-4-氧代-4-(2,4,6-三苄氧基-3-氯苯基)-2-丁烯酸乙酯(C)的合成
于50 ml圆底烧瓶中,加入0.58 g (1.23 mmol) 1-(2,4,6-三苄氧基-3-氯苯基)乙酮(B),溶于15 ml无水甲苯中,形成均相溶液,冷却至0 ℃。向溶液中分批次加入73.6 mg (1.84 mmol)氢化钠(含量为60%),搅拌5 min后,逐滴加入183 μl(1.35 mmol)草酸二乙酯。相应的混合液加热回流11 h,反应完毕,冷却至室温,溶剂减压浓缩,加入1 N的盐酸溶液酸化。乙酸乙酯萃取,饱和食盐水洗,有机相合并,无水硫酸钠干燥,浓缩,柱层析纯化,得513 mg淡黄色油状物(C),收率73%。1H NMR (400 MHz,Acetone-d6),δ:7.33~7.56 (m,15H),7.03 (s,1H),6.65 (s,1H),5.36 (s,2H),5.27 (s,2H),5.08 (s,2H),4.31 (q,J= 7.2 Hz,2H),1.31 (t,J= 7.2 Hz,3H);ESI-MS:calculated,573.03;found,573.43,[M+H]+。
2.3 5-(2,4,6-三苄氧基-3-氯苯基)异噁唑-3-羧酸乙酯(D)的合成
于50 ml圆底烧瓶中,加入325 mg (0.57 mmol)2-羟基-4-氧代-4-(2,4,6-三苄氧基-3-氯苯基)-2-丁烯酸乙酯(C),溶于15 ml乙醇中,随后加入49.6 mg (0.71 mmol)盐酸羟胺。反应液加热回流4 h,冷却至室温,过滤,滤饼依次用乙醇、水、乙醇洗涤,得263 mg淡黄色油状物(D),收率81%。1H NMR (400 MHz,Acetone-d6),δ: 7.33~7.56 (m,15H),7.06 (s,1H),6.87 (s,1H),5.36 (s,2H),5.28 (s,2H),4.95 (s,2H),4.40 (q,J= 7.2 Hz,2H),1.37 (t,J= 7.2 Hz,3H);ESI-MS:calculated,570.16;found,570.43,[M+H]+。
2.4 5-(3-氯-2,4,6-三羟基苯基)异噁唑-3-羧酸乙酯(E)的合成
于50 ml圆底烧瓶中,加入261 mg (0.458 mmol)5-(2,4,6-三苄氧基-3-氯苯基)异噁唑-3-羧酸乙酯(D),溶于15 ml无水二氯甲烷中,氩气环境、搅拌状态下,于0 °C逐滴加入2.75 ml 1 M的BCl3溶于二氯甲烷的溶液,滴加完毕,反应液升至室温搅拌3 h。反应完毕,于0 ℃搅拌下向反应液逐滴加入饱和的NaHCO3溶液,溶剂减压蒸除,水相用乙酸乙酯萃取,无水硫酸钠干燥,浓缩,柱层析纯化,得87.8 mg淡黄色固体(E),收率64%。1H NMR (400 MHz,Acetone-d6),δ:9.18 (s,1H),9.08 (s,1H),8.57 (s,1H),6.93 (s,1H),6.37 (s,1H),4.42 (q,J= 7.2 Hz,2H),1.38 (t,J= 7.2 Hz,3H);ESI-MS:calculated,299.66;found,298.17,[M-H]-。
3 结果与讨论
3.1 保护和脱保护策略
目前常用的酚羟基保护基主要有乙酰基、三甲基硅基或苄基。鉴于乙酰基和三甲基硅基不能耐受酸性环境,故选择苄基作为酚羟基保护基。此外,苄基的脱保护条件是需要考虑的另一因素。苄基常用的脱保护条件为H2/Pd/C,但此条件极易脱除分子中的氯原子,因此并不适合此反应。BCl3/DCM体系可在较为温和的条件下脱除苄基,且不会影响分子中的氯原子,因此最终选择BCl3/DCM脱除苄基保护基。
3.2 合成5-(2,4,6-三苄氧基-3-氯苯基)异噁唑-3-羧酸乙酯(D)的反应条件优化
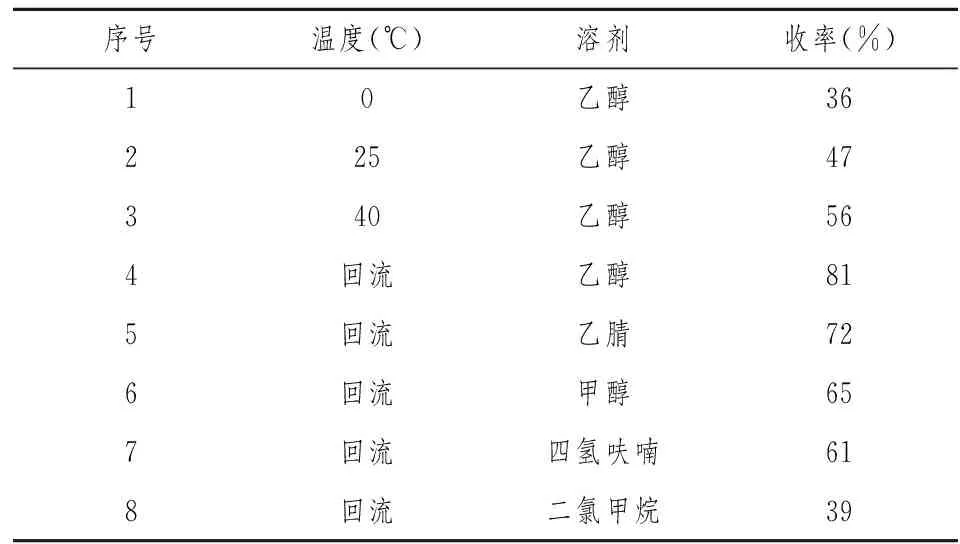
表1 反应因素对D收率的影响
如表1所示,我们对环合反应进行了反应条件优化。首先考察了反应温度,发现反应随着温度的升高,收率也逐渐提高,当反应采用回流温度时(即78 ℃),温度最高,达到81%;此外,我们对溶剂体系也进行了考察,发现采用其他溶剂,如乙腈、甲醇、四氢呋喃、二氯甲烷在各自的回流温度下反应时,均不如乙醇作为溶剂时的收率高。
3.3 5-(2,4,6-三苄氧基-3-氯苯基)异噁唑-3-羧酸乙酯(D)的结构解析
从中间体D的1H NMR数据可以看出,δ 7.33-7.56为苄基苯环上的15个H;δ 7.06为含氯原子的苯环上的H;δ 6.87为异噁唑环上的H;δ 5.36、5.28、4.95分别为苄基-CH2-上的两个H,均为单峰;δ 4.40为末端酯基-CH2-上的两个H,因受邻位耦合的影响(J为7.2 Hz),裂分为四重峰;δ 1.37为末端酯基-CH3上的三个H,裂分为三重峰。
以1-(3-氯-2,4,6-三羟基苯基)乙酮(A)为起始原料,经苄基保护、缩合、分子间环合得到5-(2,4,6-三苄氧基-3-氯苯基)异噁唑-3-羧酸乙酯(D)。对合成中间体D的反应条件进行了优化:以乙醇为溶剂、在回流温度下反应时收率最高,达到81%。最后经BCl3/DCM体系脱除苄基保护基得到了四氢苯并噻唑类小分子药物的关键中间体E。四步反应总收率达到23.1%。此合成方法具有原料简易可得、反应条件温和、收率较高等一系列优点,不仅适合多酚羟基苯基取代的异噁唑的合成,也为后续四氢苯并噻唑类衍生物的合成奠定了基础。
