DNA-PKcs抑制剂在肿瘤治疗中的研究现状
2018-10-31王傲雪马景昕
王傲雪,马景昕
(1.大连医科大学附属第二医院 皮肤科,辽宁 大连 116027;2.大连医科大学基础医学院 细胞生物学教研室,辽宁 大连 116044)
1 DNA-PKcs
DNA依赖性蛋白激酶催化亚单位(DNA-PK catalytic subunit,DNA-PKcs),属于磷酸肌醇-3-激酶相关蛋白(PI3K-related kinase,PIKK)家族,是一种丝氨酸/苏氨酸蛋白激酶。PIKK家族有4个成员:DNA-PKcs、ATM、ATR和mTOR,DNA-PKcs主要与DNA双链断裂的非同源末端连接(non-homologous-end-joining,NHEJ)修复有关[1](图1)。DNA-PKcs由prkdc基因编码,进化上保守,几乎所有哺乳动物细胞都表达。DNA-PKcs蛋白有4128个氨基酸残基,主要含有一个催化结构域、DNA结合结构域和Ku结合结构域。DNA-PKcs主要定位在细胞核,能使自身和多个下游靶点如XRCC4和DNA连接酶IV等发生磷酸化。
DNA损伤不能修复是肿瘤形成的主要原因。肿瘤中DNA-PKcs相关的变化主要有prkdc基因突变及DNA-PKcs表达和活性的改变,而且不同肿瘤组织中其活性的变化也不尽相同。研究发现,结直肠癌和胶质瘤等活性增强,乳腺癌和宫颈癌中活性降低,食道癌和淋巴瘤中活性增强和降低的报道都有[2-5]。增高的DNA-PKcs活性对于肿瘤来说是有利的。首先增高的活性使癌前细胞对DNA损伤的NHEJ修复能力提高,细胞逃脱死亡的命运。但这种修复易出错,引起DNA结构变化,促进肿瘤的形成。其次,肿瘤治疗中常用的DNA损伤性化疗药物和放疗发挥作用的主要机制就是造成DNA分子的致死性的双链断裂,进而诱导肿瘤细胞的死亡。而DNA-PKcs活性的增高能在一定程度上抑制细胞死亡,对放化疗产生耐受。此外,放化疗治疗后肿瘤组织中存活的细胞往往是DNA-PKcs活性高的细胞,它们对治疗不敏感,这也是疗效不好和预后差的原因。因此DNA-PKcs是肿瘤治疗的一个潜在靶点,采用它的抑制剂来增强放化疗的效果是一种新的策略。
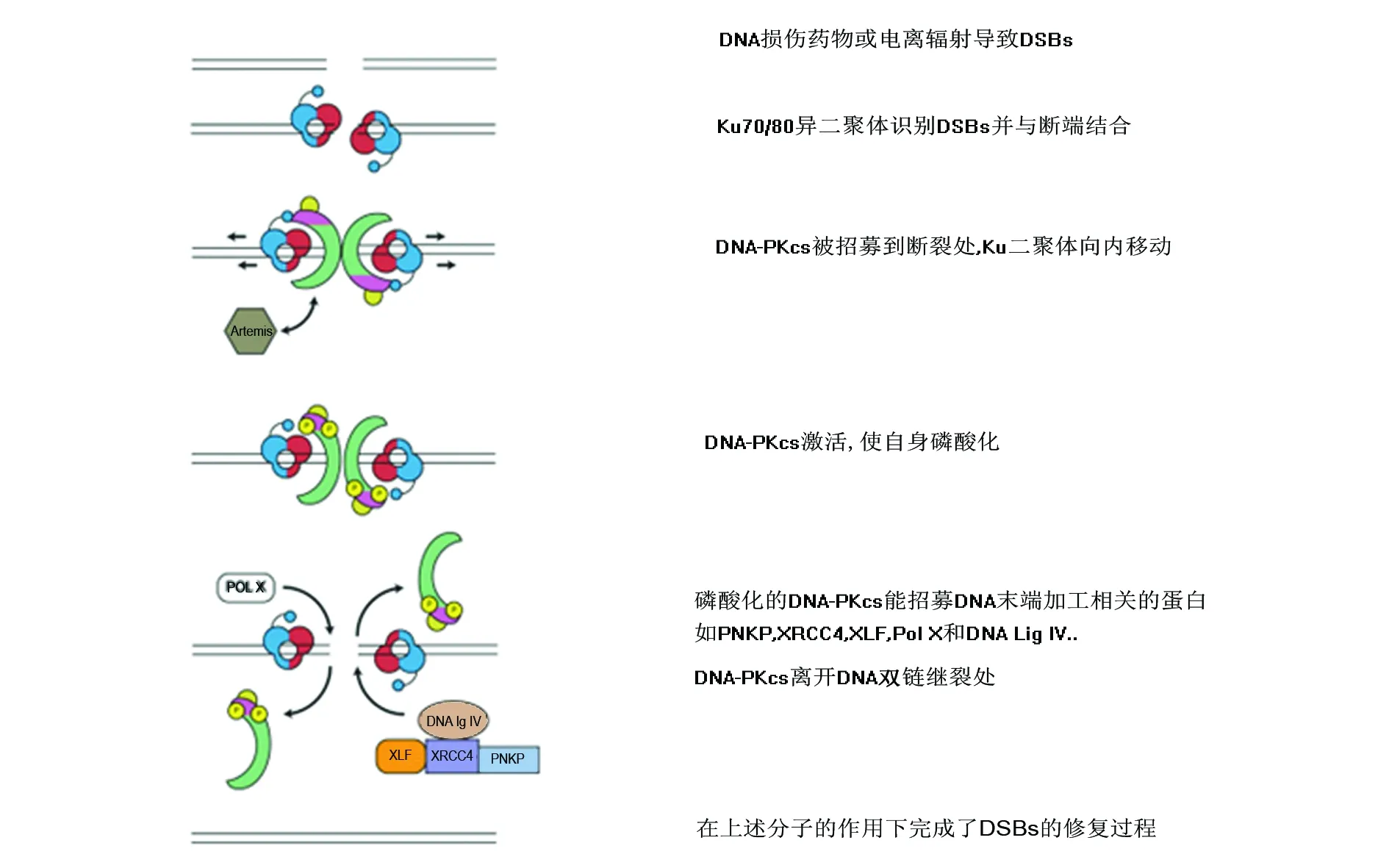
图1 DNA双链断裂的非同源末端连接修复过程中DNA-PKcs的作用[1] Fig 1 The role of DNA-PKcs in the non-homologous-end-joining (NHEJ) repair of DNA double-strand breaks (DSBs)[1]
2 DNA-PKcs的抑制剂
2.1 渥曼青霉素(wortmannin)
渥曼青霉素(图2A)也称SL-2052或KY-12420,是真菌代谢产生的一种天然类固醇。它可以通透细胞,与PI3K的110 kD催化亚单位 p110结合, 不可逆地非竞争性抑制 PI3K的激酶活性,IC50为4.2 nmol/L。较高浓度的渥曼青霉素能抑制DNA-PK的活性(IC50=16 nmol/L),能与DNA-PKcs蛋白ATP结合位点中的Lys3751共价结合或使Lys802发生不可逆的烷化,进而影响了磷酸根的转移。此外渥曼青霉素也能抑制ATM (IC50=150 nmol/L)和MLCK(IC50=200 nmol/L)等。
博来霉素是肿瘤治疗中的常用药,能引起DNA双链断裂,渥曼青霉素能显著增强它致V79中华仓鼠细胞中DNA损伤的作用。仓鼠、小鼠、鸡和人来源的DNA-PK功能正常的细胞在渥曼青霉素预处理后对电离辐射的敏感性均增高[6]。多种肿瘤细胞对辐射的敏感性也能被渥曼青霉素上调。RT112和MGH-U1是两种对X射线辐射耐受的膀胱癌细胞系。前者固有表达高水平的DNA-PKcs,而后者DNA-PKcs的表达水平低。渥曼青霉素能上调前者对X射线辐射的敏感性,但不改变后一种细胞的敏感性[7]。因此人肿瘤细胞中DNA-PKcs表达的水平可能是一种预测DNA-PKcs抑制剂用作放疗增敏剂是否有效的指标。这两种细胞中p53的表达情况也存在差异,MGH-U1中P53为野生型而RT112中p53存在纯合性突变。肿瘤中p53突变是常见的,将P53和DNA-PKcs的表达情况结合起来在选择治疗策略时更有价值。渥曼青霉素(25 μmol/L)能促进1,4苯醌诱导的p53非依赖性的人早幼粒白血病细胞HL60的凋亡[8]。

A: 渥曼青霉素; B: LY294002; C: NU7026; D: NU7441; E: KU-0060648; F: VX-984; G: M3814;H: CC-115图2 各种DNA-PKcs抑制剂的结构式Fig 2 Structural formulas of various DNA-PKcs inhibitors
渥曼青霉素的结构非常复杂,溶解度差,体内毒性大,对DNA-PKcs抑制作用缺乏特异性。同时其分子中存在一个高反应性的C20碳,导致半衰期很短只有10 min。这都限制了它的临床应用。随着特异性更好、效能更高的DNA-PKcs抑制剂被发现,渥曼青霉素在医学研究中主要作为PI3K抑制剂应用。
2.2 LY294002
LY294002(图2B)是一种含吗啉结构化合物,能以一种竞争性、可逆性方式与DNA-PKcs的激酶结构域结合。LY294002是非特异性的DNA-PKcs抑制剂(IC50=1.5±0.2 μmol/L),也能抑制PI3K的激酶活性(IC50=1.4~2.3 μmol/L),是泛PI3K/mTOR抑制剂。LY294002可以渗透细胞,在溶液中比渥曼青霉素稳定。
体外研究发现,LY294002能诱导肿瘤细胞中明显的核固缩和减少细胞质体积,显著抑制结肠癌、卵巢癌、黑色素瘤和骨肉瘤细胞的增殖[9-10]。体内实验也证实它能抑制多种肿瘤异种移植物的生长,但这主要是通过抑制PI3K活性施加的。与渥曼青霉素相比,LY294002会引起细胞内钙离子浓度的显著变化,会对细胞内其他活性造成不同的影响。如果相关研究与细胞内钙离子浓度密切相关,那么应用LY294002时就要更加谨慎。2008年研究人员发现LY294002能通过抑制DNA-PKcs的活性来使HeLa细胞对放疗变得敏感[11]。肾细胞癌是最常见的肾脏恶性肿瘤,确诊时多已发生转移,预后不好。研究发现,肾细胞癌组织中DNA-PKcs在转录和蛋白水平均高于瘤旁组织,肾细胞癌细胞系A498和786-9及原代培养的肾细胞癌细胞中DNA-PKcs的表达均高于非癌肾小管上皮HK-2细胞。LY294002能抑制上述癌细胞的增殖并诱导其凋亡,而对HK-2细胞没有类似的作用[3]。LY294002单独使用能引起人骨肉瘤细胞系U2OS和MG-63的增殖抑制和凋亡,联用时能上调这些细胞对盐霉素的敏感性。值得注意的是,LY294002或盐霉素单独使用或联用均不影响非肿瘤性成骨细胞OB-6细胞的活力[12]。这说明LY294002既有抗肿瘤活性,也有放化疗的增敏剂的作用。
LY294002的作用多样,也能抑制mTOR的活性,影响自噬体的形成和增强栎精的抗丙肝病毒活性等。LY294002存在清除率高、体内毒性大和对DNA-PKcs选择性差等问题,研究中不用作DNA-PKcs的抑制剂。但是它在制备更加有效和高选择性DNA-PKcs抑制剂中起到助力作用。在它的基础上研究人员找到了NU7026、NU7441和KU0060648等更好的DNA-PKcs抑制剂。
2.3 NU7026
研究人员以LY294002的结构为基础,合成了包括NU7026(图2C)在内的11种小分子,NU7026对DNA-PK的抑制能力约为LY294002的6倍(IC50=0.23 μmol/L)。NU7026也称LY293646,是一种竞争性、高选择性的DNA-PKcs抑制剂,对ATM和ATR没有抑制活性。尽管它也能抑制PI3K,但它对DNA-PKcs的选择性比PI3K高出60倍。与LY294002不同,它通常被称作DNA-PKcs特异性的抑制剂。
体外研究表明NU7026能增强电离辐射或DNA损伤药物的作用。中华仓鼠V3细胞存在DNA-PKcs缺陷,NU7026不能影响电离辐射对它的损伤。V3YAC细胞是携带人DNA-PKcs基因人工酵母染色体转染的V3细胞,NU7026能抑制它的生存,增强电离辐射对它的毒性作用。但NU7026并不影响电离辐射对V3细胞的损伤作用。这提示NU7026的作用与DNA-PKcs有关。在5~50 μmol/L范围内NU7026对正常肺成纤维细胞MRC-5没有细胞毒性,而对肺癌A549细胞的毒性显著,说明它是安全和有效的。NU7026预处理30 min后,X射线或碳离子照射对A549细胞的杀伤作用显著加强[13]。研究人员用γ射线反复照射历时6个月制备出对辐射耐受的肺癌细胞系A549R,采用该细胞系进行实验发现NU7026能显著增强γ射线照射的毒性[14]。此外NU7026 能显著调高CH1人卵巢癌细胞的辐射敏感性。上述结果表明NU7026能显著增强肿瘤细胞对辐射的敏感性。
多种化疗药物对肿瘤细胞的杀伤作用也能被NU7026增强。NU7026能增强伊达鲁比辛、柔红霉素、阿霉素、依托泊甙、安丫啶和米托葱醌等的生长抑制作用,但不能增强喜树碱或胞嘧啶阿拉伯氨酸的作用。NU7026能增强白血病治疗中多种药物的细胞毒性,如慢性淋巴细胞性白血病(CLL)中应用的苯丁酸氮芥耐药性的产生可能与其诱导的DNA-PKcs活性增高和DNA损伤修复加快有关。研究发现,NU7026会使CLL细胞系I83和原代培养的CLL细胞对苯丁酸氮芥变得敏感。2017年10月世界卫生组织国际癌症研究机构公布的致癌物清单中,苯丁酸氮芥出现在一类致癌物清单中,因此在肿瘤治疗中其用量的增加存在着致癌的风险。NU7026能增强低浓度苯丁酸氮芥的治疗作用,同时又能避免其致癌作用。临床上多种实体瘤对新型的嘧啶类抗肿瘤代谢药物吉西他滨反应良好,但是胰腺癌的反应性不理想。有报道证实NU7026能显著上调吉西他滨对胰腺癌细胞系的毒性作用,促进其凋亡[15]。在结直肠癌细胞中NU7026能显著提高mTOR抑制剂WAY-600的促进凋亡作用[16]。NU7026不会影响非肿瘤性的成骨细胞OB-6的活力,但能引起人骨肉瘤细胞系U2OS和MG-63增殖的抑制和凋亡,同时NU7026能上调盐霉素对骨肉瘤细胞的杀伤作用,这表明NU7026的安全性和有效性较好[12]。TRAIL能选择性杀伤肿瘤细胞,它的小分子抑制剂TIC10能有效杀死各种肿瘤细胞,是一种新的有前途的抗肿瘤制剂。NU7026对非癌的HL-7702肝细胞和原代人成体肝细胞的增殖没有影响,但能抑制两个肝癌细胞系(HepG2和Huh-7)及两种原代肝癌细胞的增殖,增强TIC10对肝癌细胞的毒性[17]。
2005年发表的小鼠体内研究,采用口服、静脉和腹腔三种方式给予NU7026,药代动力学分析后发现腹腔给药的方式最好,每天给药4次,剂量为100 mg/ kg即达到放疗增敏的效果。WAY-600诱导的抗结肠癌细胞异种移植物活性在联用NU7026的时候是增强的[16]。肝癌组织中过表达的DNA-PKcs与肝癌的发生、不良预后有关,可能是预测治疗反应性和生存情况的候选生物标记[4]。研究人员将HepG2细胞注入裸鼠,建立了肝癌异种移植物模型。口服TIC10后,肿瘤在裸鼠中的生长显著抑制。腹腔给予NU7026后,TIC10的抗肿瘤活性显著增强。而单独使用Nu7026未见显著的毒性,只产生了较弱的肿瘤抑制[17]。提示NU7026可以作为放疗增敏剂使用,有望成为抗肿瘤治疗的一种手段。NU7026的药代动力学特点决定了其在临床应用中需要处理的时间过久,有效药物浓度难以达到等问题,因此尽管它具有高选择性和较好的抑制效果,但是要用于临床必须优化。
2.4 NU7441
研究人员采用液相多重平行合成的方法,制备了第6,7和8位的芳香取代物的铬酮文库,然后把它们作为DNA-PKcs抑制剂来进行筛查,发现NU7441(图2D)的IC50为14 nmol/L。尽管它也能抑制PI3K的活性,但IC50是5 μmol/L,可见NU7441对DNA-PKcs的抑制效能和选择性都很好。
培养的细胞中,NU7441抑制DNA-PKcs后,阿霉素和电离辐射诱导的DNA双链断裂不能修复而持续存在。NU7441能上调DNA-PKcs为野生型的结肠癌SW620, LoVo和V3YAC细胞对辐射和依托泊苷的敏感性,但在DNA-PKcs存在缺陷的V3细胞中则没有这种作用。NU7441的对放化疗的增强作用是由于抑制了DNA-PKcs的活性。两种人胶质瘤细胞系M059-Fus1和M059J细胞系中DNA-PKcs功能存在差异,前者功能正常而后者缺陷。NU7441只增强阿霉素和辐射对前一种细胞的损伤[18]。NU7441在约为NU7026浓度1/10时能增强辐射对在人T淋巴细胞白血病MOLT-4细胞的杀伤作用[19]。人乳腺癌细胞系MCF-7、 MDA-MB-231和T47D中DNA-PKcs的活性在辐射后增高,非细胞毒性浓度NU7441能抑制这种变化,增加细胞对电离辐射和阿霉素的敏感性。值得注意的是,代表三阴性乳腺癌的MDA-MB-231细胞系中NU7441的致敏作用最强,提示NU7441或许能用于改善这种难治性乳腺癌的治疗现状[20]。放疗是肝癌治疗的主要手段,但是肝癌细胞修复放射性损伤的能力很强,因此需要找到能增强放疗效果的药物。NU7441能抑制HepG2细胞的增殖,并且能进一步增强60Coγ射线产生的损伤,可能是肝癌治疗中一种潜在的放疗增敏剂[21]。碳离子属于高线性能量传递的重离子,剂量控制及射程精确,定位性能好。射程末端的释放能量集中,对周围健康组织损伤小,正逐步替代传统的光子放疗。研究发现,NU7441增强了A549和H1299肺癌细胞X射线和碳离子辐射的敏感性,而且对碳离子的增敏效果更好[22]。此外肺癌中NU7441与拓扑异构酶抑制剂氨柔比星和伊立替康联用能对A549细胞的增殖产生协同抑制作用。提出NU7441和拓扑异构酶抑制剂联用可能是非小细胞肺癌的一种有前途的治疗手段[23]。鼻咽癌的体外研究也证实NU7441作为放疗敏化剂来应用的可能性[24]这些体外研究都提示它有应用价值,需要体内实验来验证。
3种方式给予小鼠NU7441后对其药代动力学进行分析。发现尽管腹腔注射的半衰期和清除率与静脉注射相似,但是生物利用度明显高于静脉注射。而口服给药的生物利用度只有约33%。因此选择了腹腔注射给药进行动物实验。测定了SW620异种移植物裸鼠腹腔注射10 mg/kg NU7441后血浆、肿瘤、肝、肾、脾和脑组织中的浓度。发现受检组织中NU7441的分布较好,而且在其从血浆清除后组织中的分布仍能维持。重要的是,在给予非毒性剂量NU7441时,在肿瘤组织中化疗致敏所需的浓度能维持至少4 h。NU7441和依托泊苷单用或联用都没有产生明显的毒性。裸鼠中SW620移植物的体积在约5.6 d的时候增加了近3倍,依托泊苷单独使用或与NU7441联用后移植物达到相同体积约需要8.3 d和11 d。可见NU7441使依托泊苷诱导的肿瘤生长延迟在毒性不增加的情况下增加2倍。人肾细胞癌组织中DNA-PKcs的转录和翻译水平均高于癌旁相对正常组织。动物实验发现在口服NU7441(10 mg/kg·d)连续3周后,人肾细胞癌细胞786-0裸鼠异种移植物的生长显著受到抑制[3]。
2.5 KU-0060648(KU60648)
KU-0060648(图2E)的作用最早是在人乳腺癌和结直肠癌中证实的。乳腺癌用的是三个细胞系:MCF7, T47D和MDA-MB-231。结肠癌用的是LoVo和SW620两个细胞系。发现它能抑制上述细胞内DNA-PKcs的自身磷酸化和PI3K介导的AKT磷酸化。因此提出它是DNA-PKcs和PI3K的双重抑制剂。KU-0060648 表现出较好的口服生物利用度和药代动力学特点。CRISPR/Cas9编辑技术提高了在精确位点上修饰基因组的能力,产生的DNA剪切主要是通过易出错的NHEJ来修复的,这限制了这项技术的应用。研究发现NU7441和KU-0060648都能减少基因编辑过程中NHEJ的频率而上调同源重组的速度,减少后续在确认含有插入的目的基因细胞时的工作量[25]。KU-0060648抑制三个肝癌细胞系HepG2, Huh-7 和KYN-2的增殖,而对于非肿瘤性的肝细胞HL-7702是没有毒性的。同时发现它对于DNA-PKcs沉默或突变的HepG2细胞仍然有细胞毒性。此时KU-0060648的作用是不依赖DNA-PKcs的,而是与PI3K-AKT-mTOR的抑制有关。因此KU-0060648抑制肝癌的作用有DNA-PKcs依赖性和非依赖性的方式两种,作用更全面,适用的细胞类型更多。但在DNA-PKcs缺陷性肿瘤中的应用要更为稳妥[5]。骨肉瘤治疗中放疗总体是无效的,有学者提出抑制DNA-PKcs等阻断DNA修复或许是可选择的放疗增敏方法。骨肉瘤细胞DNA-PKcs的蛋白水平比非癌的成骨细胞高。KU-0060648显著上调了143B和U2OS两个骨肉瘤细胞系的放射敏感性,此时细胞中DNA-PKcs的自我磷酸化显著下调,这表明它是能抑制骨肉瘤细胞中DNA-PKcs的[26]。为评价KU-0060648抗结直肠癌的潜能,采用两个细胞系HCT-116和HT-29及原代培养癌细胞进行研究。发现nM水平的KU-0060648能有效抑制人结肠癌细胞的增殖,触发凋亡。但是没有影响正常的结肠上皮细胞[27]。这表明KU0060648对于骨肉瘤、肝癌和结直肠癌等来说可能是一种有前景的放疗增敏剂。
给予携带SW620和MCF7异种移植物的小鼠无毒剂量的KU-0060648后,肿瘤异种移植物中它的浓度可以维持在体外实验中证实能抑制癌细胞生长和发挥化学增敏作用的浓度至少4 h。KU-0060648单用能延迟MCF7异种移植物的生长,并且在SW620和MCF7异种移植物中将依托泊苷诱导的肿瘤生长延迟提高到4.5倍,此时并没有将依托泊苷毒性增高到不可接受的水平[28]。在人肝癌组织中DNA-PKcs的表达是显著上调的,腹腔注射KU-0060648能显著抑制HepG2细胞移植物在裸鼠中的生长[5]。
2.6 IC化合物
早在2000-2005年之间,ICOS公司和Array生物学制药公司就发现,一些苯酚相关类化合物能抑制DNA-PKcs的活性,包括IC60211、IC86621、IC87361和IC486241等(图3)。IC60211是一种芳基吗啉,对激酶有中等强度的抑制作用。在保留芳基吗啉结构的基础上对它进行改良找到了许多DNA-PKcs的选择性抑制剂,它们在<50 μmol/L的浓度时都是没有毒性的。IC86621是IC60211的一个甲基酮衍生物,虽然不是活性最强的,但适于和易于合成,而且较稳定。IC86621结构简单,它能抑制DNA-PKcs和PI3K的激酶活性。IC87361则是一种吗啉-类黄酮,对DNA-PKcs的抑制作用比PI3K的催化亚单位p110β高50倍。要达到对放疗致敏效果则需要IC87361持续作用至少4小时,而且在循环中它很快被清除,生物利用度低,这是它应用于临床的主要障碍。IC486241能增强阿霉素或顺铂对乳腺癌细胞的毒性[29]。另一项研究中 Davidson等[30]发现IC486241能与抗结肠癌药伊立替康协同起作用,对结肠癌细胞进行杀伤。
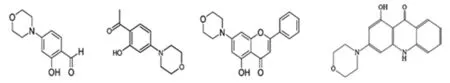
图3 苯酚相关类化合物IC60211、IC86621、IC87361和IC486241的结构式Fig 3 The structural formula of phenol-related IC Series
2.7 VX-984
VX-984(图2F)是一种氘化药物。氘也称重氢,是一种氢的稳定形态同位素,具有更强的分子键。氘化可以改变药物的多种特性如形状、大小、靶标结合力、吸收、分布及毒性。氘化能延长半衰期,减少服药次数或给药剂量,在保持药物原有活性和选择性的同时使药物更稳定。默克公司从Vertex 制药公司购买了VX-984癌症项目并在A549肺癌细胞中的进行体外实验。发现它能抑制DNA-PKcs的活性,而且作用明显强于其他PI3K家族成员[31]。采用非肿瘤性星形胶质细胞和T98G胶质瘤细胞分析发现,VX-984能抑制DNA-PKcs的活性,这种影响在肿瘤细胞中显著强于正常细胞[32],因此对于非肿瘤细胞的影响小,安全性好。
VX-984的研究主要是在胶质瘤中展开。与DNA-PKcs为野生型的M059K胶质瘤细胞相比,DNA-PKcs存在缺陷的M059J胶质瘤细胞对辐射更敏感。而导入含有DNA-PKcs基因的8号染色体能降低M059J的对辐射的敏感性[33]。体外实验证实渥曼青霉素和NU7026等都存在药代动力学不理想、生物利用度低和半衰期短等问题[34]。单独给予氘化药物VX-984不影响DNA-PKcs的活性,但它能显著抑制X射线照射后两种胶质母细胞瘤细胞U251和NSC11中DNA-PKcs活性的增高,辐射产生的DNA双链断裂不能被修复,细胞死亡增加[35],提示VX-984是放疗的增敏剂。体内实验发现,X射线照射后U251和NSC11细胞的小鼠原位肿瘤异种移植物中DNA-PKcs活性增高。如果在照射前半小时和照射后4 h喂饲溶解在乳化剂5%甲基纤维素中的VX-984后,移植物中DNA-PKcs的磷酸化水平降低,表明VX-984绕过了血脑屏障并且在脑内达到了有效浓度。VX-984单独处理负荷U251或NSC11移植物小鼠并没有影响总体生存,但VX-984与放疗联用后放疗引起的肿瘤生长抑制作用和促小鼠生存的作用都增强了[35]。可见,VX-984优先对肿瘤细胞发挥放疗增敏剂的作用,具有很好的安全性。VX-984稳定性好,并且能通过血脑屏障,是一种很有临床应用前景的DNA-PKcs抑制剂。
2.8 M3814
M3814(图2G)又称MSC2490484A,是默克公司自主研发的一种口服DNA-PKcs抑制剂,不但抑制效能高而且特异性好。为探讨M3814在实体瘤和慢性淋巴细胞性白血病治疗中的应用,2014年在健康志愿者中进行了I期临床研究。随后观察了M3814与致DNA损伤疗法如放化疗联用的效果。2016年默克公司公布了实验结果,M3814在临床前研究体系中是有活性的,而且在人肿瘤的小鼠模型中能增强辐射对异种移植物生长的抑制作用[36-37]。随后,在2017年公布了它的结构[38]。
体外实验通常是在约20%氧浓度条件下进行的,而正常组织中检测到的氧水平大约是3~7%,肿瘤组织中甚至低于2%。因此我们常规实验中细胞所处环境的氧是偏高的[39]。有研究证实氧张力降低后放疗的杀伤能力也下降[40],因此进行相关实验时应该是低氧的。为此研究人员采用了低氧的实验条件,首次发现M3814与辐射联用后能增强辐射对两个非小细胞肺癌细胞A549和NCI-H1437的杀伤能力[41]。结果提示DNA-PKcs抑制剂的治疗窗口可能比原来预想的要宽,因为大多数放疗耐受细胞都处于一种缺氧的小环境中。此外还发现,缺氧条件下M3814对碳离子作用的增强效果比正常氧条件下增高50%~60%,提示它在缺氧条件下具有更高的效能。通过像M3814这样的DNA-PKcs抑制来逆转缺氧肿瘤的放疗耐受表型是比较有前景的,值得进一步临床前和临床研究。
2.9 CC-115
CC-115(图2H)是一种有效的双重DNA-PKcs和mTOR抑制剂,IC50分别为 13 nM 和 21 nM。它也能抑制PI3K和PIKK家族的多种成员,但抑制能力很弱。体外研究发现CC-115 能抑制人前列腺癌PC-3细胞的增殖[42]。体内研究发现CC-115 表现出较好的药代动力学类型,在小鼠、大鼠和狗中口服的生物利用度分别为53%, 76%和100%。在较低的剂量如0.25 mg/kg, 0.5 mg/kg和1 mg/kg,每天两次给药或1 mg/kg每天1次给药时,CC-115使前列腺癌体积分别缩小46%, 57%, 66%和57%。同时发现给药后的1小时和3小时即表现出显著的DNA-PKcs抑制作用,并且在24 h内能维持这种作用[42]。除了前列腺癌,CC-115的抗肿瘤作用也在慢性淋巴细胞性白血病(CLL)中展开研究。B细胞受体通路能指挥B细胞恶性肿瘤进入淋巴组织,使肿瘤细胞能够接触必要的微环境而得以生存。临床上使用的伊布替尼和艾代拉里斯都是针对该通路的靶向药,后一种已经获FDA批准用于治疗复发性CLL、滤泡性淋巴瘤和小淋巴细胞性淋巴瘤。但在应用过程中发现,CLL细胞会对它们产生获得性耐药,这与DNA损伤修复有一定关系。研究发现,CC-115能抑制CLL细胞中DNA损伤的修复,并引起休眠的CLL细胞发生Caspase依赖性的死亡。尽管CC-115也能诱导健康B细胞的死亡,但IC50为0.93 μM,高CLL细胞的IC50近一倍。在CLL患者中,CC-115减少其淋巴结病的发生。体内体外实验都提示CC-115适合于临床药物开发[43]。
此外,OK-1035、SU11752和NK314也都对DNA-PKcs具有一定程度的抑制作用,但存在抑制能力弱、药代动力学类型不理想,作用特异性差等问题,因而相关研究很少。
3 DNA-PKcs抑制剂的临床研究
尽管许多分子被证实能抑制DNA-PKcs活性,目前相关的临床研究主要集中在以下四种。一项LY294002的相关临床研究是关于一种水溶性的小分子药物前体SF1126的。SF1126中含有与含RGD的四肽相结合的LY294002,能增加肿瘤细胞中LY294002的浓度,具有潜在的抗肿瘤和抗血管生成的活性。研究是在复发和难治性神经母细胞瘤患者中进行(NCT02337309)。但已提前终止。VX-984在美国临床试验网中也仅有一项研究(NCT02644278),是在晚期实体瘤和淋巴瘤中分析VX-984与聚乙二醇脂质体阿霉素联用的效果已按计划完成。M3814的研究有三项。在小细胞肺癌中与依托泊苷和顺铂联用的Ib/II期研究(NCT03116971)因没有招募足够的研究对象和研究内容的变化而提前终止。晚期实体瘤中M3814与分割放疗或顺铂联用的研究正在招募(NCT02516813)。另一项在晚期实体瘤和CLL中进行的单独M3814用药的I期临床已于2018年4月按计划完成。CC-115相关临床研究有三项。正在招募阶段的是在去势治疗耐受的前列腺癌患者中CC-115与恩杂鲁胺联用的I期临床试验(NCT02833883)。已完成招募,研究正在进行中的是晚期实体瘤和血液恶性肿瘤相关的I期临床研究(NCT01353625)。还有一项处于II期临床研究阶段(NCT02977780),主要对包括CC-115在内的几种可能用于胶质母细胞瘤药物的安全性和有效性进行研究。
4 展 望
放疗和DNA损伤药物在肿瘤治疗中十分重要,它们抗癌的机制都与诱导肿瘤细胞发生致死性的DNA双链断裂有关,这种损伤主要是通过NHEJ来修复的,因此NHEJ中起关键作用的DNA-PKcs激酶的抑制剂具有增强放化疗敏感性的作用。目前已证实多种小分子能抑制DNA-PKcs活性,但是效能和选择性各有不同。尽管大量的体外研究证实DNA-PKcs有很好的抗癌活性和放化疗增敏作用,但在进入体内研究阶段出现了药代动力学不理想、效能低、选择性差等问题,限制了它们的临床应用。目前一些DNA-PKcs进入临床研究阶段,相关的研究将会为该类药物的研发奠定基础,值得关注。
