帕金森病理机制常谈常新:多巴胺能神经的氧化性死亡
——ferroptosis和oxytosis*
2018-10-30段文君李怡芳栗原博何蓉蓉
段文君,李怡芳,栗原博,何蓉蓉
(1.暨南大学广东省疾病易感性及中医药研发工程技术研究中心 广州 6125052;2.暨南大学广东省中药药效物质基础和创新药物研究重点实验室 广州 6125052;3.暨南大学药学院,中药及天然药物研究所 广州 6125052)
帕金森病(Parkinson's disease,PD)也称为震颤麻痹,是一种以黑质致密部神经元的死亡为特征的慢性进行性神经退行性疾病。临床上常用行为功能的异常作为诊断PD的指标,主要包括动作迟缓、僵硬、静止性震颤、姿势不稳,且随病情发展,不对称地由一侧发病发展为双侧发病。PD主要的病理特征为中脑黑质致密部多巴胺能神经细胞的损伤和神经元内路易小体的出现,这也是临床诊断和治疗PD患者的重要依据。
目前研究认为凋亡(apoptosis)、程序性坏死(necroptosis)和自噬性死亡(autophagic death)等死亡方式都参与了多巴胺能神经元的死亡[1]。但是,这些死亡方式都不足以解释其病理进程和机制,深入研究发现这些死亡方式都存在一个共同特征,即:多巴胺能神经元细胞的脂质过氧化损伤。最新的研究也表明,中脑黑质致密部多巴胺能神经元的脂质过氧化是PD重要发病机制[2],而真正可能反应细胞脂质过氧化的细胞死亡方式或许是近些年新发现的ferroptosis(暂译名:铁死亡)[3]。这种细胞死亡的形态学特征表现为线粒体嵴消失,线粒体变小,膜密度增加,细胞变小变圆且相互之间分离,生物化学特征表现为脂质活性氧(reactive oxidative species,ROS)累积,谷胱甘肽(gluta⁃thione,GSH)耗竭,谷胱甘肽过氧化物酶4(glutathione peroxidase 4,GPx4)抑制,多不饱和脂肪酸释放增加等,因其在形态学、基因学、生物化学特征上与传统的死亡方式均有明显的不同,并且依赖细胞内的铁,而不是其他金属,所以命名为ferroptosis[4,5]。此外,大约30年前在神经细胞里发现了一种涉及GSH减少、ROS产生增加、脂氧合酶活化和钙内流的细胞死亡途径[6],并最终被命名为oxytosis(暂译名:氧化凋亡)[7]。这两种死亡方式在特征和信号调节等方面都有着高度相似性,最新的研究指出ferroptosis和oxytosis可能是同一种细胞死亡方式[8]。在这篇综述中,我们讨论了这两种高度相关的程序性细胞死亡途径,并总结了目前它们与神经元死亡相关的研究进展。
1 多巴胺能神经元退行性死亡是帕金森病的主要病理机制
PD患者的神经病理学诊断需要检测黑质部位神经元的损伤或死亡、路易小体和路易神经突的出现[9]。路易小体是嗜酸性的包涵体,电镜下观察其有一个致密核,周围环绕一圈苍白色辐射状的纤丝样光晕[10]。错误折叠的α-突触核蛋白(α-synuclein,α-syn)是路易小体的辐射状光晕的重要组成成分,存在于突触前末端,负责维持突触囊泡储备池的正常功能,调节神经递质的释放[11-13]。正常的α-syn主要以单体构型(14kDa)存在,并处于单体和聚合体的平衡状态[14]。在特定应激条件下,例如氧化应激[15]、翻译后修饰[16,17]、蛋白水解[18,19]、脂肪酸的浓度[20,21]、磷脂和金属离子[22]都可能诱使它形成稳定的多聚体和其他不同构型。在PD患者和转基因α-syn动物模型的脑部中多个区域发现α-syn的异常分布和聚集,被认为是多巴胺释放异常的原因之一[23,24]。体内和体外的实验数据都证明原纤维化的低聚体是α-syn毒性形式之一,它能改变生物膜的通透性引起钙离子内流导致细胞死亡,通过损伤线粒体引发溶酶体泄露,损伤微管,以及与轴突转运相关蛋白相互作用,导致突触功能异常[25-27]。最近的研究认为,持续的多巴胺能神经元损伤依赖于低聚化的α-syn向纤维化的转变[28]。此外,过表达的α-syn抑制多巴胺合成过程的限速酶,酪氨酸羟化酶的基因表达和蛋白活性[29]。
氧化应激一直是解释帕金森病多巴胺能神经元损伤的基石。从20世纪八十年代开始,越来越多的文章报道称,无论诱发神经元死亡的诱因是什么,ROS的形成都是关键的一步,是神经元损伤或死亡的较为本质的原因[30,31]。在帕金森病患者的黑质致密部甚至全身都能够检测到脂质氧化、蛋白质氧化和DNA的氧化[31-33]。黑质部位铁含量、钙离子通道活性和蛋白水解功能的改变,以及α-syn的堆积和突变蛋白的出现,都与氧化应激有关[34-37]。尽管通过控制氧化应激可以缓解体内和体外的帕金森病模型的病程发展,但是单纯依靠清除ROS的治疗手段在临床研究中的应用暂时尚未获得成功。目前对于ROS的来源还存在争议,或来自于神经元,或来自神经胶质,但可以达成一致的是ROS的产生始于线粒体或内质网[30]。基于线粒体的神经细胞死亡可以解释家族遗传性以及零散发生的帕金森病。MPTP的活性代谢物MPP+、鱼藤酮和番荔枝素都通过抑制线粒体呼吸链复合物I表现出神经毒性作用,在帕金森病患者体内也能检测到这种抑制作用,同时三羧酸循环中酮戊二酸脱氢酶也有损伤,表明线粒体呼吸链复合物I受损是帕金森病的发病类型之一[38-40]。家族发病的遗传学研究显示,α-syn、parkin、PINK1、DJ-1和LRRK2的表达或活性异常都与线粒体功能障碍有关[9,34,41]。
在多巴胺神经元细胞膜上,多巴胺神经递质代谢旺盛,产生神经毒性代谢物多巴胺醌和6-羟基多巴胺(6-OHDA)[42],这极有可能是多巴胺能神经元对氧化应激非常敏感的主要原因[43]。多巴胺醌是主要代谢产物,环化形成无色多巴胺色素并缓慢氧化为神经黑素单体。醌类化合物通过将蛋白质巯基和胺基团烷基化以及在ROS和GSH二硫化物的存在下促进蛋白质氧化来传递毒性[44]。这些蛋白质的生物化学异常将导致功能障碍,并破坏细胞膜的完整性,最终导致细胞死亡。6-OHDA也是线粒体复合物I和IV的有效抑制剂,并且可能被铁进一步氧化成反应性半醌[38]。此外,6-OHDA的代谢还产生过氧化氢,其参与与亚铁离子的Fenton反应,通过形成羟基自由基来诱导氧化应激,导致DNA加合物形成、脂质过氧化、膜完整性丧失以及细胞凋亡[45]。
2 多巴胺能神经元ferroptosis死亡方式可能是帕金森病的新病理机制
Ferroptosis是在开发治疗肿瘤的药物的过程中新发现的一种细胞死亡方式[46]。2012年Dr.Brent R.Stockwell及其团队首次发现小分子erastin能诱导选择性致死原癌基因Ras的细胞发生一种明显不同于凋亡、坏死和自噬的铁依赖性的细胞死亡方式,主要通过诱导由铁依赖的脂质过氧化累积而杀伤细胞或损害细胞功能[3,5],具有线粒体皱缩和膜密度增加的独特细胞死亡形态[3,47]。Ferroptosis可以被两类小分子化合物诱导:一类是谷氨酸胱氨酸转运体xc-系统(一种最初由Bannai和Kitamura[48]提出的胱氨酸谷氨酸反向转运蛋白)抑制剂;另一类是GPx4抑制剂[49]。除了这些小分子物质以外,还有许多药物(例如:索拉菲尼、青蒿素及其衍生物)也可以诱导ferroptosis[49]。活化的线粒体电压依赖性阴离子通道、丝裂原激酶MAPK、上调内质网应激和抑制细胞膜xc-系统是诱导细胞ferroptosis的主要原因[47,50,51],其主要机制在于诱导脂质过氧化物的生成和ROS的堆积。而这种堆积很大程度上能够被一些铁螯合剂(去铁胺、去铁敏)和脂质过氧化抑制剂(ferrostatin、lipeoxstatin、齐留通)所抑制[3,52,53]。GPx4、热休克蛋白HspB1,核因子E2相关因子Nrf2能够通过抑制铁离子摄取和抑制ROS生成来负向调节ferropto⁃sis进程[54,55]。另一方面,NADPH氧化酶、p53等则能够通过促进ROS的生成和抑制SLC7A11(胱氨酸、谷氨酸转运离子通道主要配体)的表达来诱导ferroptosis的发生[56,57]。同时,多种代谢途径如甲羟戊酸、转硫以及谷氨酰胺途径等,对ferroptosis的调控也起着至关重要的作用[58,59]。Ferroptosis已经被发现参与到很多人类疾病的病理和生理进程之中,包括肿瘤、神经毒性、神经退行性疾病、急性肾损伤、药物诱导的肝脏损伤、肝脏/心脏缺血再灌注损伤、T细胞免疫等[49]。
2.1 Ferroptosis在帕金森病中研究进展
Ferroptosis的主要特征为依赖铁离子的脂质过氧化损伤诱导的细胞死亡,同时其调节机制主要围绕GSH的合成及GSH过氧化物酶活性来展开。在目前已经研究的多种PD模型中都发现可能有ferroptosis参与其中,ferroptosis在MPTP诱导的小鼠PD模型中发挥作用,给予ferrostatin-1能有效的保护MPTP和鱼藤酮所带来的多巴胺能神经元死亡[60]。但是结合之前的科学研究和ferroptosis的调控机制,我们不难发现,谷氨酸盐神经毒性、大脑的铁负荷以及GPx4缺失所诱导的脂质过氧化可能都会以诱导ferroptosis的方式影响PD病理进程。总结PD研究中可能涉及ferroptosis调节机制,能够对ferroptosis在PD中的作用研究形成借鉴。
在诱导ferroptosis的体外实验中,给予小分子erasin可以抑制胱氨酸/谷氨酸逆向转运蛋白(xCT),进而阻断细胞对胱氨酸的摄取并引起细胞发生ferroptosis[3]。胱氨酸在细胞内转化为半胱氨酸,这是GSH合成的限速前体。当xCT被抑制时,胱氨酸摄取减少,导致GSH合成水平降低;GSH过氧化物酶如GPx4消耗GSH并清除过氧化氢和磷脂氢过氧化物[4]。GSH也是LIP中亚铁离子的天然配体[61],LIP是神经元内相对游离的铁离子的交换池。GSH在LIP中与铁离子的结合可以防止亚铁被氧化,一方面保持铁离子的溶解度(三价铁是高度不溶的),另一方面抑制亚铁作为把潜在氧化物如羟自由基变成过氧化氢的催化剂。谷胱甘肽的耗尽释放铁离子从而产生羟自由基,由此引起ferroptosis中所观察到的脂质过氧化。
除了erasin外,RSL3通过结合GPx4并使其失活而触发ferroptosis[4]。因此,给予erasin和RSL3均可引起脂质过氧化[3,4]。此外,采用插入诱变筛选了两个涉及脂质代谢的基因,酰基辅酶A合成酶长链家族成员4(ACSL4)和溶血磷脂酰胆碱酰基转移酶3(LpCAT3),是依赖于GPx4失活的ferroptosis所必需基因[62]。继而有研究证实ACSL4是前ferroptosis基因,而且是GPx4失活后脂质过氧化发生的关键基因[63]。此外,敲除AC⁃SL4表达减少了磷脂酰乙醇胺以及花生四烯酸和肾上腺素的的量,降低了细胞对ferroptosis敏感性[63]。
Ferroptosis是铁离子依赖的细胞死亡方式,同时PD患者或模型的大脑中也发现了大量铁离子的聚集。目前的研究表明,铁负荷能够以多种方式诱导多巴胺能神经元细胞的死亡:①通过Fenton反应诱导大量的ROS生成[64];②激活MAO-B活性,诱导多巴胺氧化和更多内源性ROS的生成[65];③多巴胺代谢产物可以与铁离子和过氧化氢反应生成神经毒性化合物,6-OHDA[66];④铁离子可以增加PD的标志物,α-syn的纤维化和聚集[67]。虽然目前还没有研究报道α-syn涉及到多巴胺能神经元ferroptosis的死亡进程,但是其与铁负荷之间的关联已早有报道。α-Syn对铁离子和亚铁离子都有较强的亲和力,而且这两种形式的铁离子在体外被证明能够加速α-syn的聚集,给予铁螯合剂后则能显著抑制这种现象的发生[68,69]。同时α-syn还可以作为铁离子的还原剂,能够将铁离子还原为二价铁离子,从而加重了神经元细胞内的铁负荷和ferroptosis所依赖的ROS的产生[68]。铁反应元件(IRE)是细胞能够调节蛋白质翻译以调节神经元铁负荷的关键位点,目前已经证明了α-syn mRNA的5’端的非翻译编码区是一个潜在的IRE[70]。这就意味着铁离子浓度的增加可能通过与IRE的结合进一步诱导神经元细胞内α-syn的表达增加来损伤神经元。在一项研究中这个观点也得到了证实,铁离子的耗竭能够减少α-syn的表达[71]。α-Syn还能与维持细胞内铁离子稳态的蛋白进行病理性相互作用,在MPP+处理的神经纤维瘤细胞中发现,抑制了在铁离子摄入和ferroptosis中其重要调节作用的蛋白(铁离子转运受体1)TfR1后,能够显著的减少α-syn的表达和聚集[72]。这些研究都能表明α-syn与细胞内铁负荷具有直接的相互作用,预示着他们在PD的病理调节中具有共同的调节途径,比如说ferroptosis。
2.2 Ferroptosis信号通路蛋白在帕金森病中的调节作用
铁螯合作用能够逆转erasin诱导的ferroptosis[3,52],表明铁离子是细胞死亡途径的必要介体。容易发生ferroptosis的细胞中上转铁蛋白受体TfR1是上调的,TfR1与转铁蛋白形成铁离子吸收的复合物,并下调参与细胞内铁离子储存的铁蛋白的表达[52]。抑制ferrop⁃tosis的药物包括ferrostatin-1、去铁酮、N-乙酰半胱氨酸(N-acetyl-L-cysteine,NAC)和 liproxstatin-1[60,73]。Ferrostatin-1和liproxstatin-1通过抑制脂质过氧化反应来防止ferroptosis[73,74]。铁螯合剂去铁酮通过抑制铁离子蓄积来逆转细胞ferroptosis死亡[60],而NAC则通过提高细胞内GSH水平来防止ferroptosis的发生[4]。
前人对ferroptosis信号通路中的关键调控因子的研究显示其有几种因子确实参与到了PD的病理进程中,铁离子在黑质多巴胺能神经元中的富集是PD的主要特征[75]。作为强还原剂,铁离子负荷能够导致大量羟基自由基的生成并诱导多巴胺的氧化,这可能造成了细胞内的氧化微环境并导致黑子多巴胺能神经元的损伤和丢失[76]。而且导致铁离子代谢失衡的基因突变常常会导致PD的发生,进一步证明了铁离子含量升高在PD病理进程中发挥关键的作用。几种涉及到细胞铁摄取和转运的蛋白质的突变也跟PD相关。Borie等人[77]的研究表明转铁蛋白的突变跟增加PD的风险有关。这些预示着铁摄取机制在这些帕金森患者中过度活跃,并导致了细胞内的铁富集。转铁蛋白受体TfR2蛋白的突变在PD中起到保护作用,可能跟其能减少细胞内铁摄取并降低铁的富集有关。神经元内的铁离子转运出去都要通过膜铁转运蛋白,淀粉样蛋白前体蛋白(APP)能够在膜上稳定膜铁转运蛋白的表达,保证细胞内铁离子的转出并减少铁累积。APP几种罕见突变以及几项AD家族史的研究表明APP的突变与PD有关[78]。血浆铜蓝蛋白能够将细胞内的二价铁转化成三价铁并以转铁蛋白的形式储存或转运出细胞,而其编码基因的突变也跟PD有关[79]。铁转出功能缺陷对PD的影响在黑质多巴胺能神经元中也得到了验证,APP表达降低会导致神经元大量丢失,并增加细胞内血浆铜蓝蛋白的表达水平。氧化应激对帕金森进程的影响已经有多种研究,脂质过氧化在PD患者的黑质多巴胺能神经元中已经被检测出来,而且其能够促进神经元的退行性病变[80]。谷胱甘肽水平在黑质多巴胺能神经元中水平显著降低也是PD病理进程中早期的标志[76]。
2.3 Ferroptosis抑制剂在帕金森病中具有潜在的保护作用
多巴胺能神经元中氧化代谢活动频率极高,但是其细胞内的抗氧化酶体活力低,且富含能够催化氧化还原反应的铁,因而对氧化应激特别敏感。其内丰富的多巴胺也能够通过酶解代谢和非酶代谢产生毒性极大的活性氧[81]。目前的研究表明,铁负荷是PD中较为容易处理的靶标。在多巴胺能前体衍生的神经元细胞系LUHMES细胞中施用ferroptosis抑制剂、铁螯合剂去铁酮和NAC,能缓解erastin诱导的细胞毒性,而凋亡和自噬抑制剂则不能起到保护作用[60]。此外,LUHMES细胞中的多巴胺毒性可以通过ferroptosis抑制剂所缓解,而增加多巴胺表达则加剧erastin毒性,暗示着多巴胺能神经元可能固有地对ferroptosis敏感。这也就解释了为什么铁和多巴胺的富集会增加多巴胺能神经元变性的脆弱性[42]。在离体器官培养种,用erastin能够显著的增加大脑纹状体内细胞的丢失,并且ferroptosis的抑制剂ferrostatin-1能够显著的缓解MPP+诱导的细胞毒性,而且在MPTP处理前24小时注射小鼠ferro⁃statin-1可以显著挽救小鼠的行为障碍和神经元损失[60],这表明在动物模型中常见的PD相关毒素引起神经变性的机制有可能是ferroptosis。在细胞培养和MPTP诱导的小鼠PD模型中给予铁螯合剂,如去铁酮等处理,会减少细胞内的ROS生成,增加多巴胺的利用率,从而改善现有的运动能力和抑制运动神经元的功能恶化[82]。在一组PD患者的研究中,给予18个月的去铁酮治疗可以显著的缓解早期帕金森症状并减缓运动缺陷的进展,在三期临床试验中能够重复这一结果,证明了铁在PD中的关键作用[83]。随后的研究观察发现,使用去铁酮六个月的PD患者大脑增加了多巴胺能神经元内GSH含量,而GSH的合成前体NAC,已经进入PD治疗的三期临床试验[84]。另外,此前发现一类格列酮的化合物能够选择性的抑制ferroptosis信号通路中参与磷脂代谢和膜磷脂合成的酶ASCL4[85]。这类化合物能够显著的减少糖尿病病人中PD的发病概率,并且其已经被发现了能够协同liproxstatin-1抑制细胞ferroptosis的发生[63],暗示着其作为PD药物开发的前景。
3 Ferroptosis和oxytosis可能是同一种非凋亡程序性细胞死亡
1988年,来自Coyle's团队的Murphy及其同事报道称,在N18-RE-105神经母细胞瘤X视网膜细胞系中,谷氨酸盐以及谷氨酸盐类似物使君子氨酸和鹅膏蕈氨酸诱导一种钙依赖的细胞延迟死亡的形式[6]。由于这种细胞的死亡类型被低胱氨酸培养基(一种GSH前体)加剧,因此推测其与细胞内GSH的消耗有关,其特征在于氧化应激的增加,并且能被亲脂抗氧化剂所抑制[86]。研究证明谷氨酸介导的对胱氨酸摄取的抑制是谷氨酸暴露和GSH消耗之间的关联性机制[86]。在这一点上,负责谷氨酸抑制性的胱氨酸摄取的转运系统与胱氨酸谷氨酸反向转运xc-系统具有高度相似之处。研究表明,这种细胞死亡的类型与GSH消耗后ROS生成的显著增加有关,随后钙内流导致最终的细胞死亡[7]。2001年,这种新的非凋亡的程序性细胞死亡被命名为oxytosis,该名称突出了这种类型的细胞死亡的特征为ROS堆积,并强调它是一种不同于细胞凋亡的程序性细胞死亡[7]。
抑制细胞对胱氨酸的摄取是大多数oxytosis的实验模式中的关键起始步骤。胱氨酸可通过四种转运系统进入细胞:兴奋性氨基酸转运蛋白(EAAT)[87]、b0,+系统(rBAT与SLC7A9的异二聚体[88])、介导天冬氨酸、谷氨酸和胱氨酸反向转运的rBAT与AGT1/SLC7A13异二聚体[89],以及xc-系统[90]。使用底物抑制剂如氨基己二酸盐、高半胱氨酸盐和使君子氨酸从药理方面抑制xc-系统[6,86]或从xCT敲除小鼠提取从基因方面抑制xc-系统的细胞[91]均可诱导细胞死亡,表明胱氨酸摄取抑制引起的xc-系统的抑制与oxytosis的发生有关。然而,除了胱氨酸饥饿或抑制胱氨酸摄取外,使用谷胱甘肽半胱氨酸连接酶(glutamate cysteine ligase,GCL,GSH生物合成中的限速酶)抑制剂丁基硫酸亚砜抑制GSH的合成,可诱导oxytosis的发生[90]。而在xCT的高表达的情况下,胱氨酸/半胱氨酸可以弥补GSH的缺失[92,93]。
Ferroptosis的诱导剂erastin[3]就是一个xc-系统抑制剂[51],并且通过由xc-系统抑制而导致的旁路半胱氨酸耗尽可以逆转erastin诱导的转录因子的变化[51,91]。从两种细胞死亡方式在脂氧合酶、GPx4、ROS这几方面的变化特性可以得出一个合理的假设:ferroptosis和oxytosis代表着非常相似(或甚至相同)形式的程序性细胞死亡。许多研究表明,ferroptosis和oxytosis的发生都依赖于脂氧合酶的活性,引起ROS大量产生并最终导致细胞死亡。尽管涉及的具体的脂氧合酶种类可能取决于细胞类型,但目前的数据均表明GSH的耗尽导致脂氧合酶的活化,继而与PEBP1相互作用并与膜(特别是细胞内细胞器膜)结合,传递脂质ROS、氧化磷脂酰乙醇胺(包括花生四烯酸或肾上腺酸),而形成细胞死亡[5,94,95]。GPx4的失活最终导致的结果与脂氧合酶活化相同:生物膜内部小叶中脂质ROS的堆积。在体外xc-系统抑制时,GSH耗尽导致脂氧合酶的活化和GPx4的失活,两者均参与了ferroptosis和oxytosis进程。另外,由于脂氧合酶的活化需要将其二价铁氧化为三价铁,因此这两种途径可能是相互关联的。磷脂过氧化氢在GPx4抑制作用下的堆积,有利于脂氧合酶的活化[5,94,96]。ROS,特别是脂质过氧化物的产生,是ferroptosis和oxytosis的细胞级联式死亡中的重要步骤。不同来源的ROS对不同细胞类型的作用可能不尽相同。线粒体来源的非脂质ROS似乎继发于脂质过氧化氢的产生,后者极有可能因脂氧合酶的活化而产生[97]。目前还不清楚脂氧合酶诱导的线粒体外膜改变是否直接诱导线粒体来源的非脂质ROS的产生。在GPx4基因缺失的MEFs细胞和谷氨酸诱导oxytosis的HT22细胞中,ferroptosis抑制剂ferrostatin-1、liprox⁃statin-1以及1,8-tetrahydryaphthyridinol衍生物能够通过捕获过氧化自由基链而保护细胞免于死亡[98],这些作用表明脂质过氧化氢是ferroptosis和oxytosis细胞死亡中重要起始因素。
有研究提到维生素E家族的生育三烯酚可通过减少oxytosis,预防包括PD在内的一些神经退行性疾病(阿尔茨海默病、亨廷顿病等),但并未作深入的机制探讨[99]。早在2007年Xu等人[100]在小鼠海马神经元HT-22细胞的谷氨酸毒性模型中发现oxytosis的产生,程序性坏死抑制剂necrostatin-1能通过上调GSH水平并减少活性氧物质的产生而发挥细胞保护作用。2018年同样是在HT-22细胞中,Hirata等人[101]发现了一种新化合物GIF-0726-r,可以防止谷氨酸诱导的细胞死亡,包括ferroptosis和oxytosis。尽管目前还没有确切的有关oxytosis在PD发病机制中的研究,但基于fer⁃roptosis和oxytosis在信号调节等方面的高度一致性,可以认为新近探讨的ferroptosis与PD发病机制的关系与oxytosis的作用差别无二。
4 中药抗ferroptosis在PD药物开发方面的应用前景
在我国,中药具有几千年的应用历史,并且大部分是安全、有效的天然药物,含有多种活性成分。最近的研究表明发现黄芩素(baicalein)可抑制胰腺癌细胞中麦角菌素诱导的 ferroptosis,与 ferrostatin-1、liprox⁃statin-1、甲磺酸去铁胺和β-巯基乙醇等ferroptosis抑制剂相比,黄芩素具有显著的抗ferroptosis活性。黄芩素可以抑制麦角菌素诱导的二价铁生成、谷胱甘肽的消耗和脂质过氧化,并且抑制erastin诱导的GPx4的降解[102]。Probst等[103]发现黄芩素可作为选择性12/15-脂氧合酶抑制剂,保护RSL3诱导的急性淋巴细胞白血病细胞的ferroptosis。在生理条件下黄芩素可能是一种很强的铁螯合剂,调节铁稳态和抑制Fenton反应[104]。黄芩作为清热泻火药,在临床上应用广泛,其中黄芩素是其发挥药效的主要物质基础,黄芩素能降低ROS、调节铁稳态、保护GPx4等,可以作为天然ferroptosis抑制剂使用。尽管迄今为止还没有研究报道黄芩素通过抗ferroptosis而发挥对PD模型的治疗作用,但基于目前发现PD模型中ferroptosis的普遍存在性,结合黄芩素在抗ferroptosis方面的比较确定的作用和机制,可以推测黄芩素在治疗PD的药物开发方面具有可观的潜力。此外,临床上已经证明能改善心力衰竭的葛根素,也被最新的研究发现能够减少erastin或异丙肾上腺素处理大鼠心肌细胞H9c2的ferroptosis,给予葛根素能够缓解铁负荷以及脂质过氧化[105]。以往的研究表明葛根素可通过多种途径缓解ROS的产生和细胞氧化损伤,如Hsp72[106]、JNK/p38 MAPK[107]、Ca2+内流[108]等,可能具有多种靶点。
Ferroptosis作为新近的热点,目前有关中药及天然产物的相关文章很少,对于目前已有报道的单体在fer⁃roptosis的调节作用的机制(如图1)还有待于进一步探讨,以期在此基础上继续发掘具有潜在ferroptosis调节作用的中药及活性成分。中药及其活性成分具有调节靶点多、结构稳定、安全性高、廉价易得的许多优势,其中不乏在抗PD方面具有乐观前景的单体。
谷氨酸(Glu)和ferroptosis诱导剂erastin可以抑制与谷氨酸反向转运相关的xc-系统(System xc-)对胱氨酸(cystine)的摄取,从而导致谷胱甘肽(GSH)耗尽,并随后抑制GSH依赖性的GSH过氧化物酶4(GPx4)。GPx4也可以直接被RSL3抑制。GPx4的抑制导致脂氧合酶(LOX)的激活,由此导致脂质过氧化氢(Lipid-OOH)可能积累在内质网中或非常接近内质网,这是产生活性氧(ROS)的起始步骤。黄芩素(Bai)则能上调GSH并降低二价铁浓度;此外,黄芩素还能通过保护GPx4来抑制ferroptosis的发生。
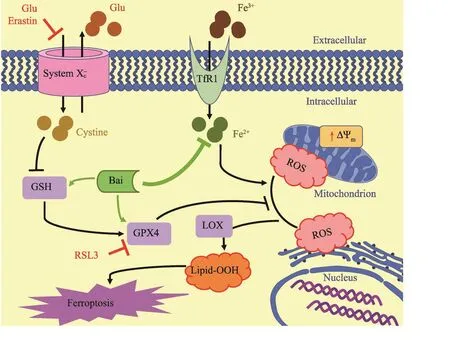
图1 Ferroptosis调节机制
5 结论
多巴胺能神经元中氧化代谢活动频率极高,但是其细胞内的抗氧化酶体活力低,且富含能够催化氧化还原反应的铁,因而对氧化应激特别敏感。其内丰富的多巴胺也能够通过酶解代谢和非酶代谢产生毒性极大的活性氧。尽管PD与多巴胺能神经元损伤的病理特征非常明显,但是其机制仍尚不清楚。Ferroptosis作为一种新被发现不依赖caspase激活而依赖于细胞内铁的细胞死亡机制,其在PD中的作用研究才刚刚开始。GSH的耗竭会诱导细胞内不稳定游离铁离子的增加,而游离的铁离子则能够通过Fenton反应增加细胞内的脂质过氧化产物。这些ferroptosis的特征在PD的病理进程中都有发现,其中包括中脑黑质神经元内游离铁的含量升高、脂质过氧化物生成、和ROS的堆积[109]。同时,应用能够抑制ferroptosis的药物也体现出了在PD治疗中的潜能,NAC和GSH合成酶的前体物,能够有效的抑制PD模型小鼠中的神经退行性病变,并在PD患者的早期临床治疗中恢复和改善运动能力[84]。铁螯合剂也被证明了能够改善动作PD模型动物的运动能力,并进入了II期临床试验[83]。最新的研究表明,ferroptosis特异性的抑制剂ferrostatin-1能够有效的减少MPTP模型小鼠中多巴胺能神经元的缺失[60]。以上的研究都显示了ferroptosis可能作为一个重要的机制参与到PD病理进程中。与此同时,证据显示ferropto⁃sis与三十年前在多种神经细胞中发现的oxytosis在信号调节等方面具有高度相似性,它们可能是同种非凋亡程序性细胞死亡的不同名称。阐明ferroptosis/oxyto⁃sis死亡方式及其机制可能为抗PD药物的研发提供重要的理论依据。
