肾透明细胞癌预后相关miRNAs的生物信息学分析
2018-10-22付军明肖招兰何富强郭君其谭建明朱凌峰
付军明 肖招兰 何富强 郭君其 谭建明 朱凌峰
作者单位:350025 福州,福州总医院泌尿外科
肾细胞癌(renal cell carcinoma)简称肾癌,是泌尿系统最常见的恶性肿瘤之一,在最近20年内发病率逐年上涨,截至2017年,其在成人恶性肿瘤中比例达到3﹪[1]。肾透明细胞癌(clear cell renal cell carcinoma,ccRCC),是肾癌最常见的病理类型,约占肾癌的70﹪~ 80﹪[2]。现阶段对ccRCC的发病机制尚未研究清楚,由于其对放化疗敏感性差,因而手术治疗仍是其首选治疗方案。近年来随着分子医学的快速发展,大量的肿瘤标志物相继被发现并应用于临床,为临床治疗提供了有力的帮助[3-5]。然而目前尚未有ccRCC的生物标志物应用于临床,因此,寻找切实有效的ccRCC肿瘤标志物显得尤为重要。
MicroRNA(miRNA)是近年来发现的一类长度约为22个核苷酸的非编码单链RNA分子,在转录后的基因表达调控中发挥关键性作用[6]。近年来已有大量报道多种miRNA参与各类型肿瘤疾病的发生与发展进程[7-9]。本研究运用生物信息学方法,从癌症基因组图谱TCGA(The Cancer Genome Atlas)数据库中下载ccRCC中miRNA表达数据,筛选出差异表达miRNA并对其进行系统性分析,为探索ccRCC发病的分子机制以及后续寻找可行的治疗靶点提供理论依据。
材料与方法
一、材料
(一)数据材料
通过GDC(https://portal.gdc.cancer.gov/) 工具下载TCGA数据库中ccRCC临床资料以及miRNA的表达数据(miRNAseq 3级数据)。选择的数据集为Kidney Renal Clear Cell Carcinoma(KIRC)。截至2018年3月,该数据集miRNA的表达数据来源于545个ccRCC样本以及71个对应的癌旁组织样本。本研究严格遵守TCGA发布的发表指导规范(https://cancergenome.nih.gov/publications/publicationguidelines)。
(二)分析软件及数据库
R软件(https://www.r-project.org/)及其附带的“edgeR”包、“Survival”包;Cytoscape软件(http://www.cytoscape.org/)及其插件“ClueGO”; Funrich软件(http://www.funrich.org/); Perl软件 ; miRNA靶基因数据库 Targetscan(http://www.targetscan.org/vert_71/)、miRDB(http://www.mirdb.org/)、miRTarBase(http://mirtarbase.mbc.nctu.edu.tw/php/index.php)。
(三)方法
利用R软件的“edgeR”包筛选出ccRCC与正常组织的差异表达miRNA,筛选标准为:|LogFC|≥ 2,adj.P ≤ 0.01。然后使用“Survival”包,运用Kaplan-Meier法对差异表达的miRNA进行生存分析,阈值设置为P≤0.05。接着利用Perl软件将生存期有意义的差异表达miRNA在三个数据库(Targetscan、miRDB、miRTarBase)中进行靶基因预测,预测结果取交集。在Cytoscape软件中对miRNA及其靶基因进行可视化分析,最后利用软件自带的ClueGO插件以及FunRich软件对靶基因分别进行KEGG(Kyoto Encyclopedia of Genes and Genomes)通路分析和GO(Gene Ontology)富集分析。
结 果
一、差异表达的miRNA筛选结果
经过对TCGA数据库中545个ccRCC样本及71正常组织样本数据的处理,一共筛选出54个差异表达miRNA,其中上调33个,下调21个(表1)。同时,利用“edgeR”包绘制出了差异表达miRNA的热图(图1)和火山图(图2)。
二、差异表达miRNA生存期分析结果
将从TCGA数据库中下载的ccRCC临床资料(共530例病人生存时间信息)进行标准化处理后,运用“Survival”包对差异表达miRNA进行生存分析,按照P≤0.05的标准,发现hsa-miR-21和hsamiR-155的差异表达与患者的总体生存率(Overall survival)具有相关性,即二者高表达时,患者总体生存率明显下降(图3)。
三、 hsa-miR-21与hsa-miR-155靶基因预测结果
将hsa-miR-21与hsa-miR-155在Targetscan、miRDB、miRTarBase和miRPath这四个数据库中进行靶基因预测,取交集结果发现共预测到129个靶基因,在Cytoscape软件中对miRNA与靶基因的关系对进行可视化(图4)。

表1 差异表达的miRNA(TOP 5)

图1 差异表达miRNA热图
四、 靶基因GO分析及KEGG通路分析结果
GO富集分析主要分为三大类:细胞组分(Cellular Component, CC)、分子功能(Molecular Function,MF)、生物过程(Biological Process,BP)。利用Funrich软件分析靶基因的MF、BP,按照P值大小排序前十名结果见图5。其中BP富集分析主要包括信号转导(Signal transduction)、细胞通讯(Cell communication)以及碱基、核苷核苷酸和核酸的代谢(Nucleobase ,nucleoside, nucleotide and nucleic acid metabolism)。KEGG通路分析结果表明靶基因主要富集在FoxO信号通路(FoxO signaling pathway)以及TNF信号通路(TNF signaling pathway)等,具体通路及相关靶基因见图6。

图2 差异表达miRNA火山图
讨 论

图3 hsa-miR-21与hsa-miR-155在ccRCC中的生存曲线
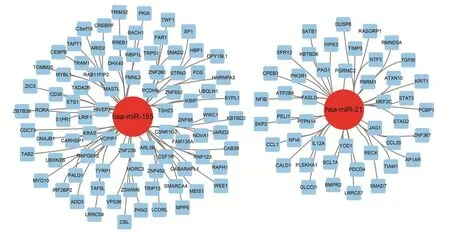
图4 miRNA-Target Genes网络图

图5 靶基因GO分析

图6 靶基因KEGG分析
在本研究中,利用TCGA数据库一共筛选出了54个在ccRCC中差异表达的miRNA,其中上调33个、下调21个。然后从中筛选出与ccRCC的预后相关的miRNA:hsa-miR-21和hsa-miR-155,并在多个数据库中预测这两个miRNA的靶基因,最后对这些靶基因进行通路富集和功能分析。通路富集分析发现这些靶基因主要与MAPK、Ras、TNF等信号通路密切相关。近年来对miRNA在肿瘤中的研究日益增多,但这些研究往往集中对在一个miRNA的单个或一类靶基因的研究[10-11]。而本研究采用的综合分析miRNA靶基因的方法,有助于对miRNA及其功能进行更全面的分析。本研究是首次利用TCGA数据库筛选出与ccRCC患者总体生存率相关的miRNA,并对其靶基因进行GO和KEGG分析,对临床上寻找ccRCC的治疗靶点具有重要指导意义。
近年来,已有大量研究表明hsa-miR-21与hsamiR-155均涉及多种肿瘤的发生发展进程。如Maachani等[12]发现hsa-miR-21在神经胶质母细胞瘤中高表达并且与肿瘤的分期分级以及病人的预后不良密切相关,在体外实验中通过敲低hsa- miR- 21的表达发现肿瘤细胞的增殖受到抑制,并且在体内实验中也证实了通过敲低hsa-miR-21能够显著的抑制肿瘤的形成。Kong等[13]研究发现在三阴性乳腺癌中,hsa-miR-155通过靶向VHL基因并影响其表达来促进肿瘤的血管生成,进而影响患者的预后情况。根据最新的文献报道,这两个miRNA均在ccRCC进程中扮演着重要角色。如hsa-miR-21在ccRCC中能够刺激上皮到间质的转化以及肿瘤发生[14],hsa-miR-155则能够影响ccRCC的增值能力以及侵袭性[15]。
GO富集分析可以深入了解miRNA的靶基因参与的分子功能和生物学过程等。在hsa-miR-21和hsa-miR-155对应的靶基因GO富集结果中,涉及到转录因子活性、信号转导以及细胞通讯等功能均与ccRCC的产生及发展有密切的联系[16-18]。KEGG通路分析结果中,诸如FoxO以及TNF等信号通路,不仅有大量文献报道这些通路与ccRCC有关[19-21],更是在其他多种肿瘤的病理过程中发挥着重要作用[22-24]。
综上所述,本研究采用生物信息学方法筛选出了ccRCC中差异表达的miRNA,通过分析hsa- miR-21与hsa-miR-155的预后生存曲线发现它们的高表达与患者的预后差有密切联系,并且二者靶基因参与的信号通路与ccRCC的发生发展有着密不可分的联系。总之,本研究对hsa-miR-21和hsa- miR-155在ccRCC中的潜在发病机制进行了深入分析,进一步对ccRCC的基础研究和临床治疗靶点的发现提供了理论指导。
