Fe2O3-CeO2光催化剂的制备及其催化性能
2018-10-11李友凤刘国清曾令玮彭振山向湘昱
李友凤, 刘国清, 曾令玮, 彭振山, 向湘昱, 王 莹, 黄 晓
(1.湖南科技大学 化学化工学院, 湖南 湘潭 411201; 2.湖南科技大学 精细聚合物可控制备及功能化应用湖南省重点实验室, 湖南 湘潭 411201)
CeO2独特的晶体结构及较高的储氧和释放氧能力,使其在催化领域备受关注[1-3]。单组分CeO2催化剂一般是通过调节CeO2的形貌和颗粒大小来提高催化剂的活性和选择性[4-6],其性能不足以将催化氧化技术推向广泛的实际应用。因此,制备高效的Ce基氧化物催化剂,增大其利用率、扩展其应用范围具有重要意义。
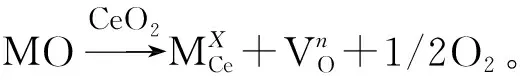
笔者主要采用水热共沉淀法和水热浸渍法制备Fe3+掺杂CeO2光催化剂,探讨共沉淀法和浸渍法对Fe2O3-CeO2结构特性和光催化活性的影响,并研究这两种方法对Fe2O3-CeO2粉末晶型、形貌及光催化活性等特性参数的作用规律。
1 实验部分
1.1 试剂和原料
六水合硝酸亚铈,质量分数为99.99%,赣州鑫正新材公司产品;硝酸铁(AR)、十二烷基硫酸钠(CP)、尿素(AR),上海国药集团化学试剂有限公司产品。蒸馏水 (自制)。
1.2 样品的制备
水热共沉淀法:首先取0.545 g十二烷基硫酸钠溶解于蒸馏水中,搅拌溶解后加入1.0 g尿素,继续搅拌使之混合均匀。再取0.868 g六水合硝酸铈和一定量的硝酸铁溶于蒸馏水中,然后将该溶液缓慢加入十二烷基硫酸钠和尿素的混合溶液中,80℃水热搅拌0.5 h,将溶液转移到100 mL聚四氟乙烯内衬的不锈钢高压水热釜中,于140℃保温 24 h。待冷却至室温,离心过滤得红色沉淀,并依次用蒸馏水和无水乙醇洗涤,将所得的沉淀于烘箱内80℃干燥10 h得到前躯体,然后该前躯体于马福炉中600℃焙烧4 h,得淡红色Fe2O3-CeO2样品。
水热浸渍法:工艺流程如上,首先制备纯的CeO2粉体。然后称取一定量纯的CeO2粉体分散于蒸馏水中,将硝酸铁加入该悬浮液中,搅拌1 h,再过滤、干燥,600℃焙烧得Fe3+掺杂CeO2样品。
实验中Fe3+的掺杂量(即Fe3+/Ce3+摩尔分数,以下同)分别为2%、3%、5%和7%。
1.3 样品的结构与表征
采用德国布鲁克AXS有限公司的D8 Advance型X射线衍射仪 (X-ray diffraction,XRD)检测所得催化剂样品的物相结构,CuKα射线,50 kV,180 mA,在2θ为10°~80°范围进行扫描。采用日本电子公司的扫描电镜 (JSM-6380LV, SEM) 观察粉体形貌结构特性。样品的织构性能在美国Micromeritics公司的ASAP2010V402A 型自动吸附仪上测定,在200℃抽真空预处理1 h后,以N2为吸附质,在-196℃下进行。用美国Perkin-Elmer公司的紫外-可见光谱仪 (Lambda 35)测量降解液的吸光度。
1.4 样品的催化降解实验
以质量浓度为10 mg/L的次甲基蓝为目标降解物,考察CeO2样品的光催化活性。取次甲基蓝溶液100 mL于250 mL 的烧杯中,以230 W紫外灯为光源,在磁力搅拌条件下,加入一定量的Fe2O3-CeO2催化剂样品。在反应开始前,先将反应溶液在暗室里磁力搅拌30 min以使催化剂表面吸附达平衡。开启光源后,每隔20 min取样,静置后经15000 r/min离心分离5 min后取上层清液,用紫外-可见光谱仪在其最大吸收波长664 nm 处测量其吸光度,计算其降解率η,见式(1)。
(1)
式(1)中,A0、A分别为降解前后次甲基蓝溶液的吸光度。
2 结果与讨论
2.1 沉淀法和浸渍法制备的Fe2O3-CeO2催化剂的物性表征
2.1.1 XRD分析
图1为不同方法制得的Fe3+掺杂量不同的Fe2O3-CeO2催化剂的XRD图谱。由图1可见,600℃煅烧后,粉末产品主要为立方萤石结构的CeO2,衍射角在10°~80°范围内有4个较强的衍射峰,对应的衍射角28.5°、33.1°、47.5°和56.4°分别为(111)、(200)、(220)和(311)晶面,其晶型与标准的JCPDS(34-0394)图谱一致[13]。共沉淀法所得的粉体为纯立方萤石结构的CeO2相,没有杂相峰出现,说明Fe3+取代Ce3+的晶格位置或均匀分布在CeO2表面。并且从图1(a)观察到,随着Fe3+掺杂量的增加,CeO2样品的结晶度提高,衍射峰强度增大。而浸渍法所得的产品,当Fe3+掺杂量为3%时,为纯的CeO2结构,没出现杂相峰;当Fe3+掺杂量为5%时,衍射角36.87°和43.07°处有微弱的杂质峰;当Fe3+掺杂量为7%时,衍射角36.87°和43.07°处有较强的杂质峰,经比对,其符合JCPDS(25-1402)型Fe2O3的衍射峰,显然当Fe3+掺杂量在5%以上时就会有Fe2O3生成。这说明浸渍法Fe3+掺杂不均匀,并且随着Fe3+掺杂量增加,衍射峰强度减小,产品的结晶度减弱。
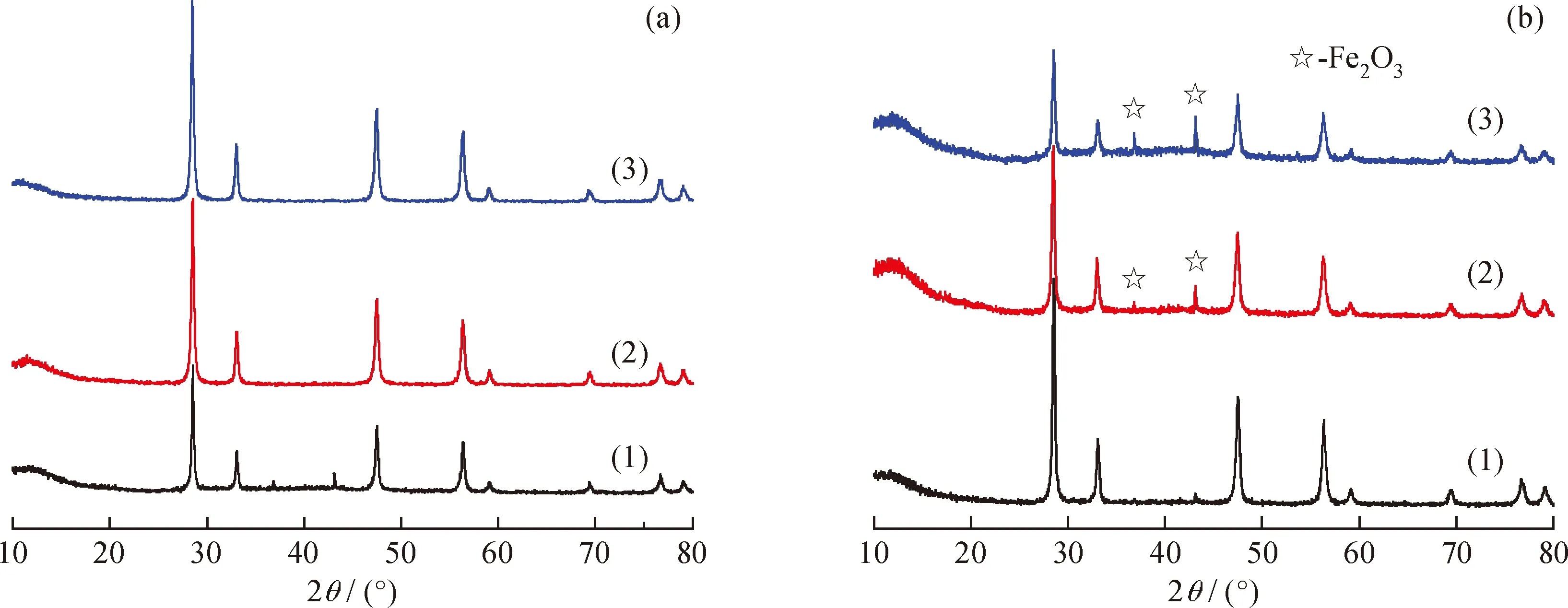
图1 沉淀法和浸渍法制得的Fe3+掺杂量不同的Fe2O3-CeO2催化剂的XRD谱图Fig.1 XRD patterns of Fe2O3-CeO2 samples prepared by coprecipitation method and impregnation method with different Fe3+ molar fractions(a) Coprecipitation method; (b) Impregnation methodx(Fe3+)/%: (1) 3; (2)5; (3)7
2.1.2 SEM分析
图2为Fe3+掺杂量为3%时,水热共沉淀法和水热浸渍法得到的Fe2O3-CeO2催化剂的SEM照片。从图2可以看出,共沉淀法所得的Fe2O3-CeO2为由小颗粒裹在一起的近球形产品,用Nano Mearsurer1.2软件计算产品直径为300~500 nm。浸渍法所得的Fe2O3-CeO2样品为不规则的小球,有些球形颗粒团聚在一起。由于十二烷基硫酸钠是一种表面活性剂,具有双亲性,在碱性条件下,随着十二烷基硫酸根离子的浓度增大,十二烷基硫酸根离子在碱式碳酸铈微晶表面的吸附量增多,晶面的结合能减小,使晶粒趋向于各向同性,颗粒形貌趋向球形,因此,高温煅烧后获得球形的CeO2样品。可能由于浸渍法得到的Fe2O3-CeO2经过两次高温烧结,样品颗粒有长大和团聚现象。

图2 沉淀法和浸渍法制得的Fe2O3-CeO2催化剂的SEM照片Fig.2 SEM photos of Fe2O3-CeO2 samples prepared by coprecipitation method and impregnation methodx(Fe3+)=3%(a) Coprecipitation method; (b) Impregnation method
2.1.3 BET分析
表1列出了沉淀法和浸渍法制备的Fe2O3-CeO2样品的比表面积、孔体积和平均孔径。从表1可以看出,600℃煅烧后,沉淀法样品比表面积和孔体积较大。这可能是由于浸渍法样品经过两次高温煅烧,导致样品颗粒长大,孔径增大,比表面积减小,这些织构特性将会直接影响到Fe2O3-CeO2的催化降解性能。
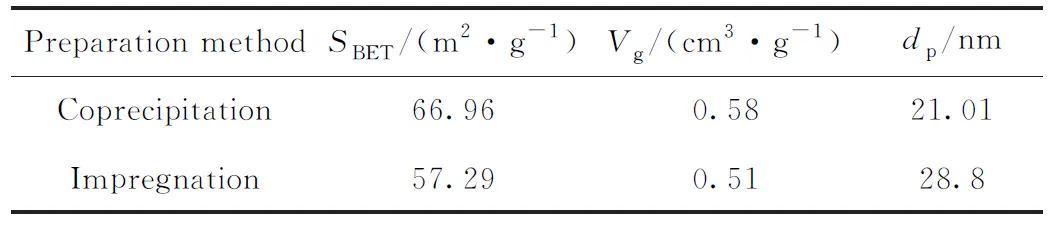
表1 沉淀法和浸渍法制得的Fe2O3-CeO2催化剂的组织结构性能Table 1 Textural performance of Fe2O3-CeO2 samples prepared by coprecipitation method and impregnation method
x(Fe3+)=3%
图3为沉淀法和浸渍法制备的Fe2O3-CeO2样品的N2吸附-脱附等温曲线图。由图3可见,两种样品均呈现出介孔材料特征的吸附-脱附等温线。由于毛细管凝聚现象,孔径大小、形状不完全均一所导致的样品有滞后环,沉淀法样品的滞后环较浸渍法明显,其样品的孔形状为瓶颈型。并且,样品的吸附曲线在相对压力(p/p0)为0.8~1.0处吸附量急剧上升,这可能是由于样品具有较大的孔径,以及比较集中的孔径分布,且孔径主要集中在介孔大小范围内。颗粒的形貌和尺寸决定了其比表面积、孔径和孔体积,从而影响样品的吸附-脱附性能。
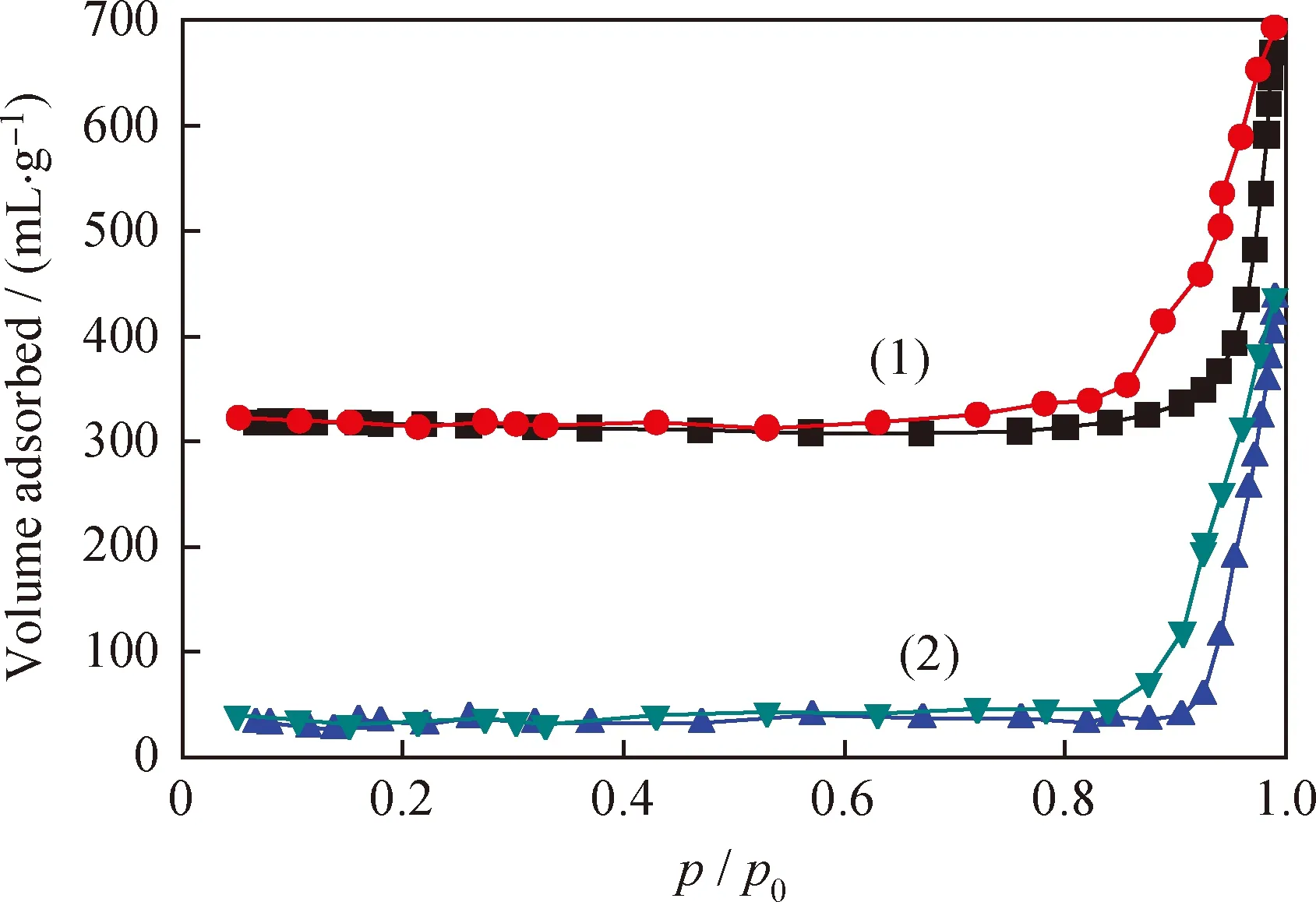
图3 沉淀法和浸渍法制得的Fe2O3-CeO2样品的N2吸附-脱附等温线Fig.3 N2 adsorption-desorption isotherms of Fe2O3-CeO2samples prepared by coprecipitation method and impregnation methodx(Fe3+)=3% (1) Coprecipitation method; (2) Impregnation method
2.2 反应条件对沉淀法和浸渍法制得的Fe2O3-CeO2光催化降解次甲基蓝的光催化性能影响
2.2.1 铁掺杂量的影响
为了研究Fe3+掺杂量对Fe2O3-CeO2样品的催化脱色效果的影响规律,称取Fe3+掺杂量为0%~7%的不同方法制备的Fe2O3-CeO2样品,取催化剂质量浓度为200 mg/L进行光催化降解次甲基蓝实验,其降解率的结果见图4。从图4可以看出,随着Fe3+掺杂量的增加,共沉淀法所得Fe2O3-CeO2样品对次甲基蓝的降解率先增大后减小,当Fe3+掺杂量为3%时,次甲基蓝的降解率最大,光催化效果最好。尽管浸渍法所得Fe2O3-CeO2样品的降解率变化趋势不如共沉淀法的明显,但其最佳光催化降解效果的样品中Fe3+的掺杂量也是3%。这是因为反应开始时,随着Fe3+掺杂量的增加,CeO2晶格内部的缺陷增多,为催化氧化提供更多的活性氧物种,大幅度提高了催化剂的反应活性;若Fe3+过量,多余的Fe3+又充当了空穴-电子的复合中心,结果使氧活性中心减少,催化降解次甲基蓝的效率反而降低[14]。
由图4还看到,在相同的工艺条件下,共沉淀法所得Fe2O3-CeO2样品对次甲基蓝的降解率最高,当催化降解时间为100 min时,其降解率达90.8%;而浸渍法所得样品,其降解率为85.2%,其值是相同催化降解时间沉淀法样品的94.6%;并且Fe2O3-CeO2样品的降解率均高于单组分CeO2的,这可能是由于CeO2光催化性能取决于活性组分Fe3+的促进作用、催化剂的晶体结构和形貌尺寸。Fe3+捕获电子形成Fe2+, Fe2O3起到了接受电子和传递电子的作用,使得光生载流子得到分离, 因而降低了CeO2的光生电子与空穴的复合几率,延长了光生载流子的寿命,增大了光电转换率,从而提高了CeO2光催化活性,所以Fe2O3-CeO2催化降解效果较纯CeO2的要好。
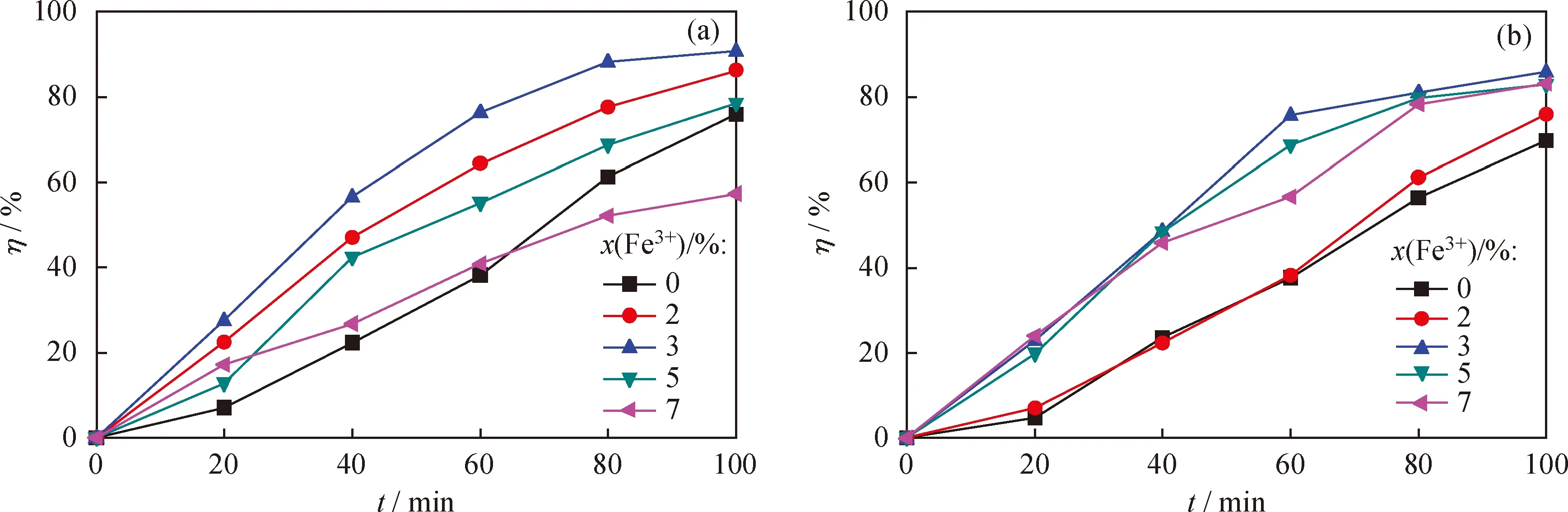
图4 沉淀法和浸渍法制备的不同Fe3+掺杂量的Fe2O3-CeO2催化剂对亚甲基蓝溶液的降解率影响Fig.4 Effect of Fe3+ amount on degradation rate of Fe2O3-CeO2 samples prepared by coprecipitation method and impregnation methodc(Fe2O3-CeO2)=200 mg/L(a) Coprecipitation method; (b) Impregnation method
催化剂CeO2为N型半导体材料,禁带宽度(Eg)为2.94 eV,低于紫外光子的能量。因此,在紫外光照下,Fe2O3-CeO2光催化剂被激发,在催化剂表面产生电子-空穴对,光致空穴和水分子作用生成强氧化性·OH,·OH与次甲基蓝反应生成CO2和H2O。掺杂的Fe3+进入CeO2晶格后,因为Fe2+/Fe3+的能级低于CeO2的导带能级,所以Fe3+有效降低了Fe2O3-CeO2光催化剂的带隙能;同时Fe3+掺杂在催化剂的晶格中产生缺陷,晶格缺陷表面容易吸附水分子,并将其氧化成大量的·OH,形成更多的氧化空穴,从而提高了光催化性能[15],因此,随着Fe3+掺杂量增加,催化降解效果提高。当Fe3+掺杂量少时,CeO2中没有足够的空穴产生·OH,光催化活性低;当Fe3+掺杂量较大时,过多的Fe3+减小了捕获载流子的空间位,使电子-空穴的复合率增加,与Fe2O3-CeO2表面的氧化反应形成竞争,催化活性降低[12]。研究发现最佳的Fe3+掺杂量为3%。
而浸渍法制得的Fe2O3-CeO2催化活性低于同条件下共沉淀法制得的Fe2O3-CeO2的,这是因为催化剂的催化活性还与其晶体结构和形貌尺寸有关。图1的XRD结果表明,浸渍法制备的Fe2O3-CeO2样品Fe3+掺杂不均匀,并且Fe3+掺杂量在5%以上时有Fe2O3生成,导致活性组分Fe3+的促进作用减弱;图2的SEM结果显示,浸渍法所得Fe2O3-CeO2样品经过两次高温煅烧后,颗粒有长大与团聚现象,导致其相应的催化降解效果降低。
2.2.2 催化剂质量浓度的影响
图5为催化剂质量浓度对Fe3+掺杂量为3%不同方法制备的Fe2O3-CeO2催化剂上次甲基蓝降解效率的影响规律。从图5可以看出,随着催化剂质量浓度的增大,催化降解率呈现先增大后减小的趋势。共沉淀法所得Fe2O3-CeO2样品的降解率随催化剂质量浓度变化比较明显,当质量浓度为100 mg/L、催化降解时间为100 min时,对次甲基蓝的降解效果最好,其降解率达97.6%;而浸渍法所得Fe2O3-CeO2产品的降解率随催化剂质量浓度变化较小,当质量浓度为50 mg/L时,催化降解100 min时降解率最高达93.4%。这可能是由于随着催化剂质量浓度的增加,催化活性中心增多,催化降解效果更好;但当催化剂质量浓度进一步增大时,屏蔽效应将降低催化剂的光能转换效率,同时催化剂对光的散射也会导致其光催化活性的下降[16]。
2.2.3 pH值的影响
实验发现,降解液在酸性或中性环境下蓝色溶液基本不褪色,在碱性条件下更有利于催化剂的光催化反应。图6为pH值对光催化效果的影响。从图6可以看出,Fe2O3-CeO2催化剂在pH值约为10.0时均达到最佳的催化降解效果,尽管光照降解初期,pH值为11.0时,次甲基蓝的降解率相对要高,但随着时间延长至100 min,两种Fe2O3-CeO2样品在pH值为10.0时都呈现出最佳的催化降解效果。Fe2O3-CeO2复合氧化物中空穴与溶液中的水分子的—OH氧化形成·OH自由基,然后·OH与次甲基蓝反应生成小分子物质、CO2和H2O[17]。·OH自由基的氧化能力是水体中存在的氧化剂中最强的, 所以溶液的pH值越高,—OH越多,生成·OH越多,次甲基蓝的降解率则越高。但是,当有大量·OH存在时,·OH又会与溶液中的H2O2反应生成O2,降低了次甲基蓝的降解率。因此,当降解液pH值为10.0时,次甲基蓝的降解率最高。
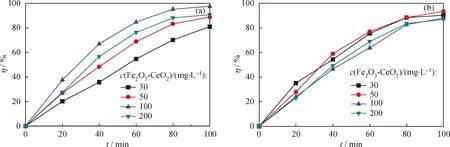
图5 催化剂质量浓度对Fe3+掺杂量为3%的Fe2O3-CeO2催化剂上次甲基蓝降解效率的影响Fig.5 Effect of catalyst mass fraction on degradation rate of Fe2O3-CeO2 with 3% Fe3+ prepared by coprecipitation method and impregnation method(a) Coprecipitation method; (b) Impregnation method
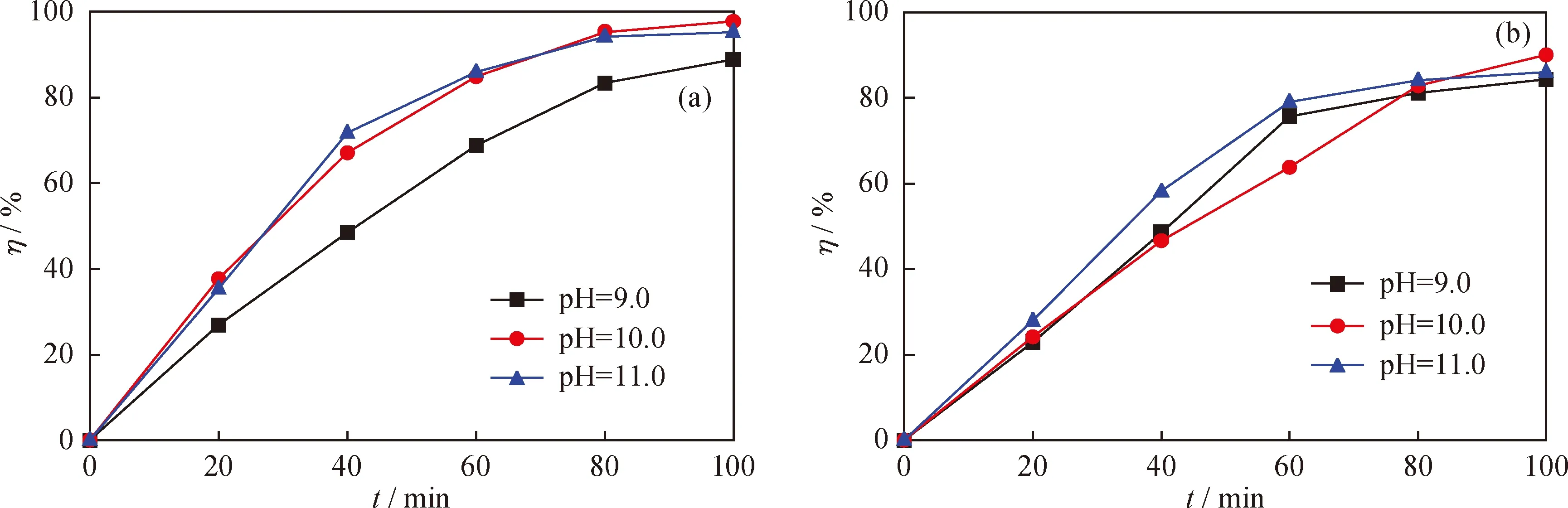
图6 降解液pH值对Fe3+掺杂量为3%的Fe2O3-CeO2催化剂上次甲基蓝降解率的影响Fig.6 Effect of solution pH value on methylene blue degradation rate by Fe2O3-CeO2 catalyst prepared following coprecipitation method and impregnation method(a) Coprecipitation method; (b) Impregnation method
2.3 不同方法制备的Fe2O3-CeO2催化剂上次甲基蓝催化降解过程的动力学研究
在工艺条件相同情况下,笔者研究了不同Fe2O3-CeO2样品降解次甲基蓝过程的动力学因素,选取共沉淀法、浸渍法与单组分CeO2样品对应的最佳降解率条件,采用最小二乘法进行线性拟合,得出次甲基蓝的光催化降解过程遵循准一级动力学方程,见式(2)。Pouretadal等[18]研究指出,多相光催化反应动力学模型均符合L-H动力学方程。
(2)
式(2)中,r为反应速率,mg/(L·min);t为反应时间,min;ρ0为次甲基蓝的初始质量浓度,mg/L;ρ为t时刻次甲基蓝的质量浓度,mg/L;Kdye为L-H吸附平衡常数,L/mg;k为表观速率常数,mg/(L·min);kapp为一级准速率常数,min-1。
图7为不同方法制备的Fe2O3-CeO2和纯CeO2催化剂降解次甲基蓝过程中ln(ρ0/ρ)-t的拟合曲线。从图7可知,共沉淀法制备的样品所得直线斜率最大,准一级速率常数kapp为14.52×10-2,比浸渍法样品增大23.9%,比纯CeO2样品升高71.8%,说明共沉淀法制备的Fe2O3-CeO2降解次甲基蓝的速率要快,催化效果要好。
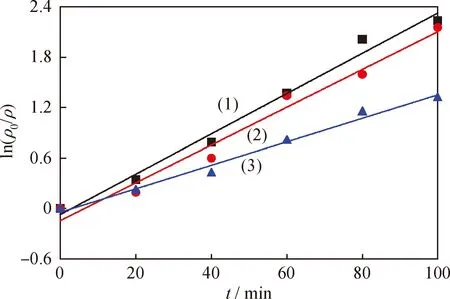
图7 不同方法制备的Fe2O3-CeO2和纯CeO2催化剂催化降解次甲基蓝过程中ln(ρ0/ρ)-t的拟合曲线Fig.7 Fitting curve of ln(ρ0/ρ)-t in the process of methylene blue degradation by pure CeO2 and Fe2O3-CeO2 catalyst prepared with different methods c(Fe2O3-CeO2)=20 mg/L; x(Fe3+)=3%(1) Coprecipitation method; (2) Impregnation method; (3) Pure CeO2
3 结 论
(1) 以十二烷基硫酸钠为表面活性剂,利用尿素水热共沉淀法合成球形的Fe2O3-CeO2,产品纯度高,为立方萤石相结构的CeO2;而水热浸渍法合成球形的Fe2O3-CeO2,当Fe3+掺杂量为5%以上时,有Fe2O3杂相峰出现。
(2) 随着Fe3+掺杂量的增大,Fe2O3-CeO2样品对次甲基蓝的降解率先增大后减小,最佳Fe3+掺杂量为3%。随着催化剂用量的增大,次甲基蓝的降解率先增大后减小,共沉淀法最佳催化剂质量浓度为100 mg/L;而浸渍法的最佳催化剂质量浓度为50 mg/L。次甲基蓝溶液的pH值直接影响到降解过程,当pH值为10.0时,次甲基蓝的降解率最高。共沉淀法所得Fe2O3-CeO2样品在最佳工艺条件下,催化时间为100 min时,对次甲基蓝的降解率达97.6%;而浸渍法所得Fe2O3-CeO2样品的降解率最高达93.4%。
(3) 多相光催化反应符合L-H动力学模型,共沉淀法样品的准一级速率常数kapp为14.52×10-2,较浸渍法样品增大23.9%,比纯CeO2样品升高71.8%。
