双稳态分子的自旋调控与功能化
2018-10-10陶军
陶 军
(北京理工大学 化学与化工学院,北京 100081)
1 双稳态分子
分子基材料的性质取决于其电子结构和几何(晶体)结构.通常情况下,分子基材料合成以后,其电子、几何结构是固定不变的,它的宏观物理性质也是不变的.但是,也有一些分子基材料合成以后,其电子结构和(或)几何结构在某些外界因素刺激时可以发生变化,而且变化前后的状态都可以稳定存在,这一类材料的分子称为双稳态分子.对于双稳态分子来说,它们的性质变化包括金属离子的电子自旋态互变、磁化强度方向互变、金属-有机分子双向电子转移、电偶极矩方向互变、结构相变等[1].双稳态分子的性质变化可以是自发的,也可以是受激发生的,而且大多数变化是可逆的.两种状态之间存在能量上的差别,与之相对应的化学和物理性质也因此而不同(图1).如果将双稳态分子的两种状态(或其宏观物理性质)看成是1和0,则双稳态分子本身或由其构成的材料可应用在纳米尺度范围的开关、存储、显示、传感等领域.
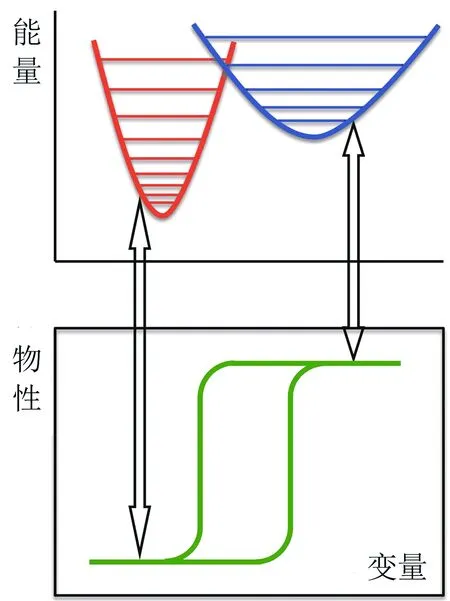
图1 双稳态分子的能量与物理性质示意图
双稳态分子的类型可以划分成两大类,根据金属离子的电子结构性质划分有自旋交叉、价态异构、金属间电子转移等[2],根据偶极矩性质划分有铁磁、铁电、单分子磁体等[3-5].在双稳态转变过程中可能伴随有结构相变,而结构相变本身也可以看成是一种双稳态,有一些结构相变是导致性质出现双稳态的重要因素.文中所述的研究领域是自旋交叉和价态异构双稳态分子材料,作者在这篇综述中总结了自旋交叉分子的自旋调控、多功能化等方面的工作.
自旋交叉(spin crossover, 简称SCO)[6]指的是金属离子的自旋态在高低(中间)自旋之间相互转变的现象,目前仅发生在3d过渡金属离子的化合物中,而且以八面体配位化合物居多.对于3d4-7构型的过渡金属离子(MnII, MnIII, FeII, FeIII, CoII, CoIII)来说,其自旋在八面体配位几何中有两种排列方式,如果配体场分裂能大于电子成对能,则金属离子处于低自旋态(low spin, 简称LS);反之,则是高自旋态(high spin, 简称HS).而四面体配位的配体场分裂能通常较小,因此很少四面体配位的3d4-7构型过渡金属离子化合物有自旋交叉性质,除非是有非常强的配体存在(如卡宾类配体)[7];五配位的3d4-7构型过渡金属离子化合物(三角双锥或四方锥)也很少发生自旋交叉行为[8];对于正方形配位几何的化合物来说,如NiII(3d8)在正方形场中是低自旋的,但可以通过轴向配位键的生成和解离来实现低自旋与高自旋的转变[9],因此也可以发生自旋交叉行为.
目前,自旋交叉的研究主要集中在二价铁(II)的化合物中,这是因为Fe(II)化合物的自旋转变伴随的性质变化比其他金属离子化合物的性质变化明显很多,从结构、性质表征到应用方面的角度来说研究都相对容易.以八面体构型的Fe(II)化合物为例,在其Tanabe-Sugano图中(图2,左)[10],Fe(II)离子的低自旋基态为1A1g,高自旋基态为5T2g,配体场强度10 Dq在10 000~20 000 cm-1范围内其基态可能发生翻转,即从1A1g到5T2g或从5T2g到1A1g转变.这种基态翻转(crossover)也就是自旋态翻转,即自旋交叉(SCO).如果自旋态翻转的同时伴随有相变发生,这种自旋交叉也称为自旋转变(spin transition, 简称ST).通常对于八面体构型的Fe(II)化合物,如果配体场分裂能小于11 000 cm-1,则该化合物基本只处于高自旋态;如果配体场分裂能大于21 500 cm-1,则该化合物基本只处于低自旋态.这两种情况都不会发生自旋交叉行为.如果化合物处在高自旋态,但配体场分裂能在11 500和12 500 cm-1之间,则可以发生高自旋向低自旋的转变;如果化合物处在低自旋态,但配体场分裂能在19 000~21 000 cm-1之间,则可以发生低自旋向高自旋的转变.因此,自旋交叉的发生与金属离子的自旋态和配体场分裂能的大小密切相关.另外,根据公式ΔG= ΔH-TΔS,可以算出在高低自旋转变时其平衡态温度T1/2= ΔH/ΔS(ΔG= 0, %LS = %HS).
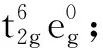
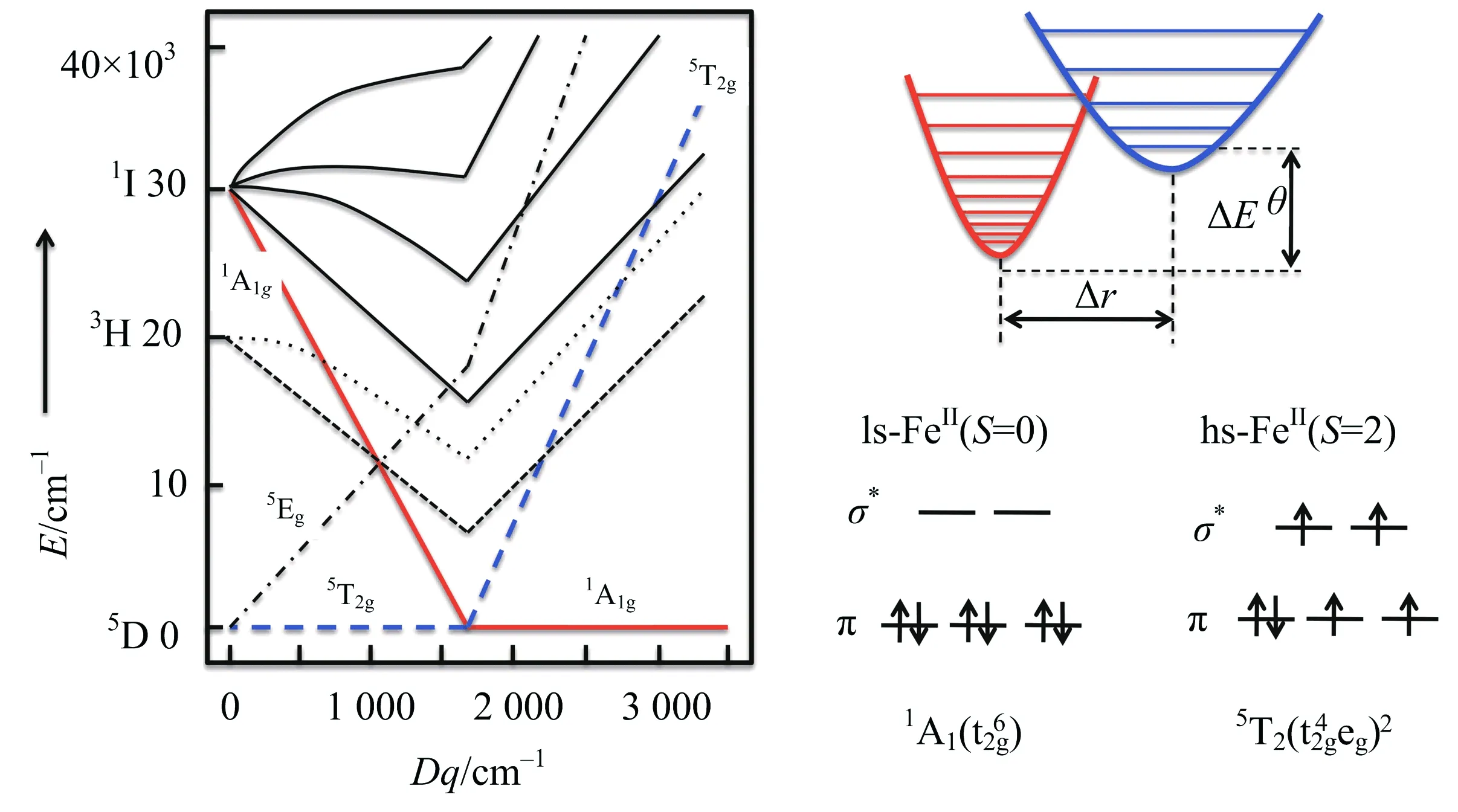
图2 二价铁离子的Tanabe-Sugano图(左)和自旋态变化示意图(右)
2 自旋调控方法
对于自旋交叉化合物来说,配体场强度合适就有可能发生自旋交叉行为,但并不是说就一定会发生,因为还有很多因素会影响到自旋交叉的发生与否.比如,晶格中如果有溶剂分子存在,则溶剂分子可以通过氢键或偶极作用影响到自旋交叉分子,使其配位场增强或减弱,从而导致自旋交叉发生或不发生.又比如,自旋交叉分子结晶时的晶型不同,即分子的堆积方式不同,可以导致分子间的弱相互作用不一样,晶格内的“化学压力”就会不同,因此有的晶型可以发生自旋交叉,有的晶型则不行,即使组成不同晶型的晶体内分子完全相同.但是反过来看,这些影响因素又可以被利用,变为我们的控制手段来实现对自旋态的调控.另一方面,通过分析自旋交叉过程中金属离子的内在性质变化,还可以发现其他调控自旋态的方法,比如光激发或压力诱导.
如图3所示[11],八面体场中的Fe(II)在低自旋时可以通过光激发的途径从基态(1A)跃迁到激发态(1T),该激发态可以快速驰豫到中间态(3T),然后再从中间态跃迁到高自旋基态或回到低自旋基态;同样,在基态高自旋时也可以通过光激发跃迁到激发态(5E),然后通过中间态跃迁到基态低自旋或回到基态高自旋.因此,可以通过光激发从低自旋快速转变成高自旋,或者进行反向快速转变.从某种意义上说,我们可以利用光学方法来控制分子自旋,使其按照要求处于低自旋态或高自旋态,或者实现其高低自旋的双向转变.从动力学角度来看,光激发诱导自旋交叉要比热诱导自旋交叉快很多.但是,由于激发态寿命通常只在纳秒数量级,因此光激发自旋交叉只能在非常低的温度下才能实现,不过近期的研究表明光激发自旋交叉的工作温度已经接近液氮温度.光激发的最大优势是可以利用不同波长的光实现自旋态的双向调控,这在某些特殊条件下具有非常广阔的应用前景.

图3 二价铁离子的激发态能级与光诱导自旋交叉
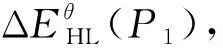
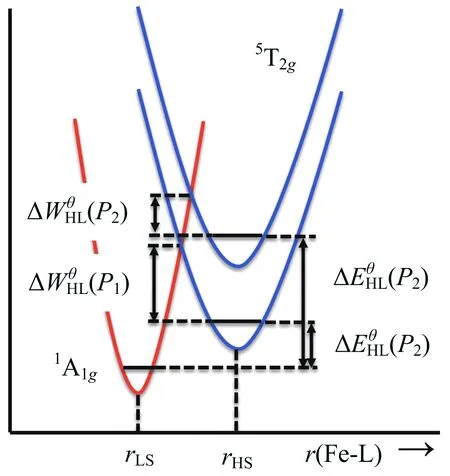
图4 二价铁离子的压力诱导自旋交叉
因此,影响自旋交叉或者控制自旋态的因素或方式很多,一个分子或体系可能受到各种因素的共同影响,故需要综合考虑.作者在研究过程中对各种影响因素综合考察,在结合他人工作的基础上,提出了自旋交叉分子电子自旋态影响因素的3个层次(图5),即分子本身的配体场强度、分子间的相互作用和外部环境.
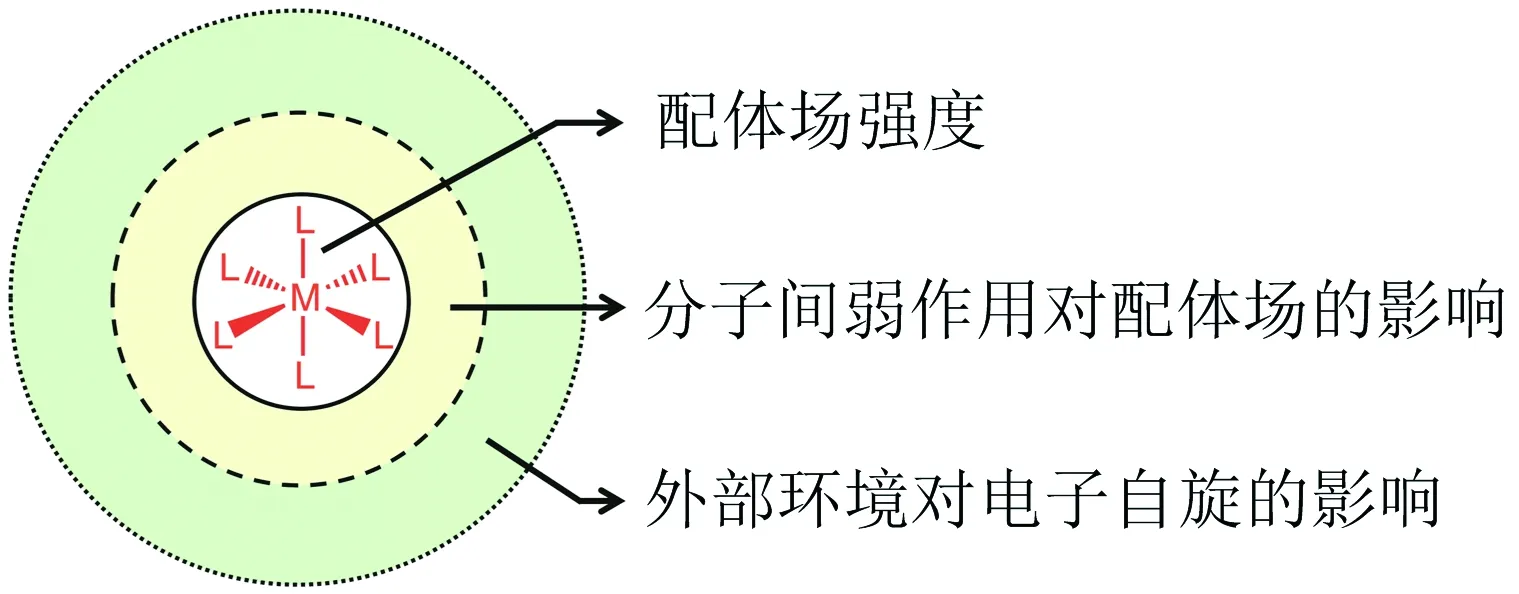
图5 自旋交叉化合物电子自旋调控策略
从上述3个层次出发,把调控自旋态的手段划分成化学调控和物理调控,如图6所示.
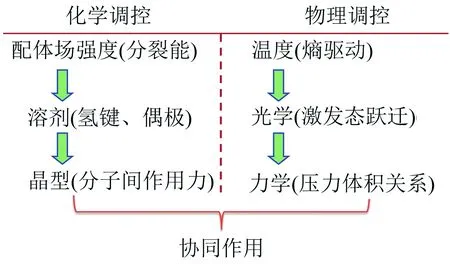
图6 作者在调控自旋交叉化合物自旋态时采取的化学和物理方法
化学调控首先是从合成的角度出发,设计合适的有机配体,使其与Fe(II)配位时具有合适的配体场强度;然后控制结晶过程中晶格溶剂分子的种类和数量,或者控制晶格中溶剂分子的可逆交换;进一步可以控制晶体的晶型,即控制分子间的堆积方式和相互作用.化学调控大多是间接的,手段相对来说比较简单,但预期的调控效果有时不佳.相反,物理调控的方法避免了设计或改变配体场分裂能,大多数情况下调控效果明显,物理方法包括温度控制、光激发、压力控制、电激发等.笔者在自旋交叉化合物电子自旋的化学与物理调控方面做了深入探究,工作进展如下.
3 自旋交叉分子的自旋调控研究进展
3.1 配体场调控
笔者课题工作是从一个文献早已报道的自旋交叉化合物[FeII(tpa)(NCS)2] (1, tpa = tris(2-pyridylmethyl)amine,图7A)开始的[13-14].文献报道化合物1有一步的不完全自旋交叉行为(图7B, 右),但是没有晶体结构.笔者课题组首先成功地培养出该化合物的单晶并测定了其结构,证实了配体tpa在配位时可提供强度合适的配体场,使Fe(II)的自旋交叉行为处在合适的温度范围内,因此得以开展以此化合物为母体的一系列自旋调控工作.比如微调配体结构,将[FeII(tpa)(NCS)2]配体tpa的其中一个亚甲基更换为—CH2CH2—,则一个配位五元环变成六元环,相应的配体场强度得到微调,使其自旋交叉的温度向低温区移动[15].如果将[FeII(tpa)(NCS)2]的NCS-更换成NCSe-(结果目前尚未发表),则配体场强度得以增强,使该化合物的自旋交叉温度可以向高温区移动.
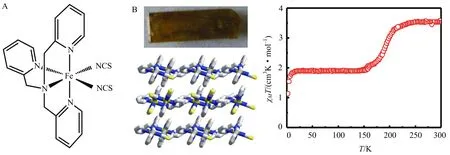
图7 用于自旋态化学和物理调控的母体化合物1(A)以及它的晶体、结构和磁性(B)
3.2 溶剂分子调控
化合物1的晶体中并没有溶剂分子,因此原本无法考察溶剂分子对其自旋态的调控,但是笔者课题组成功地将甲醇分子从气态直接引入到晶格当中,甲醇分子与晶格中化合物1的部分分子之间形成氢键,使其配体场强度和部分未形成氢键的化合物1分子的配体场强度出现差异.因此,甲醇分子进入晶格后出现了两种金属中心,两种金属中心的自旋交叉温度出现明显的差异.换句话说,可以人为地通过甲醇分子来控制化合物1分子的自旋态,而且仅凭肉眼观察颜色变化就可以判定甲醇分子改变了金属的自旋态(图8)[16].

图8 化合物1 分子自旋的甲醇分子调控
更重要的是,不仅是甲醇分子,只要存在一定蒸汽压的有机溶剂,其分子都可以从气态直接进入化合物1晶体的晶格,而且不同溶剂分子对化合物1分子的自旋态调控效果是不同的.因此,从某种程度上来说,可以利用化合物1的晶体来检测环境当中是否有某种有毒的有机溶剂分子.另一方面,对于上述含溶剂分子的晶体,笔者课题组还成功地实现了这些晶体对溶剂分子的可逆、气固相交换(图9),交换前后的Fe(II)中心的自旋态也能够可逆地相互转变[17].与通常的在结晶过程中不可控制的引入溶剂分子不同,笔者课题组是在后期可控地引入溶剂分子,从而可控地利用溶剂分子来调控化合物1的自旋态.

图9 交换溶剂分子可逆地调控化合物1 分子的自旋
3.3 晶型调控
化合物1在生长晶体过程中,很快以晶型I的形式结晶出来,如果将晶型I的晶体压碎放在某种混合溶剂中,则其形状和颜色在20 d左右的时间内连续地发生变化,依次为橘色长棒状、紫色小块状和黑色块状(图10).同一个化合物的晶体长出不同的颜色和形状预示着可能存在不同的晶型,通过单晶结构分析发现这4种晶体确实是化合物1的不同晶型.磁性测试表明其自旋转变的温度从pI到pIV依次升高,这就意味着分子的配体场愈来愈强.由于化合物分子本身并没有发生变化,这种配体场强度的变化显然是由分子间相互作用力变化引起的.如果将化合物1的分子看成是一个建筑模块的话,在不同的晶型中模块的堆积方式是不同的.这种不同在结晶过程中通常比较容易实现,但是笔者课题组的不同晶型是通过连续的、固态到固态的转变得到的,而且这种晶型转变是100%的[18].所以说可以通过晶型来调控自旋交叉化合物的电子自旋,在晶体转变到某种颜色时就可以停止进程,获得具有特定自旋交叉性质的晶型.这种晶型的连续转变实际上是晶体表面的分子在溶剂诱导下首先发生排列方式的变化,表面变化然后引起晶体内部分子的排列方式也跟着变化,是一种溶剂诱导的晶型转变.
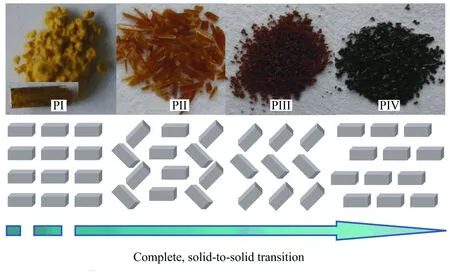
图10 化合物1 的连续晶型转变导致自旋交叉性质发生变化
在晶型的转变过程中,其动力学过程可以用Ostwald规则来解释(图11)[19].化合物1的晶型I是一个动力学亚稳态,在表面有溶剂分子作用时可以降低其晶型转变的活化能,从而逐步转变成热力学稳定态的晶型.
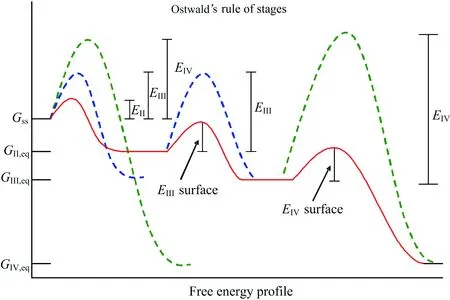
图11 化合物1 的连续晶型转变能量关系示意图
3.4 温度调控
自旋交叉化合物的自旋态转变Gibbs自由能为ΔG= ΔH-TΔS,自旋转变过程的ΔH值通常很小,而从低自旋向高自旋转变其ΔS值是增加的,是熵驱动的过程,受温度的影响很大.在自旋态发生50%转变时的温度是T1/2= ΔH/ΔS,因此升高温度可以使平衡向高自旋态移动,降低温度可以使平衡向低自旋态移动.即利用温度就可以调控分子的自旋态.以化合物[FeII(tpa){N(CN)2}]4(2, 图12 inset)为例,该化合物为正方形的四核自旋交叉化合物,在高温时4个Fe(II)原子均为高自旋,当温度降到260 K左右时,一条对角线的2个Fe(II)原子转变为低自旋;当温度继续降到175 K以下时,另一条对角线的2个Fe(II)原子也转变为低自旋[20].升温过程自旋态的转变是可逆的,这就是温度对自旋态的调控.
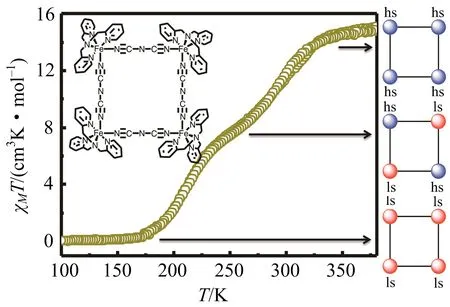
图12 化合物2 自旋态的温度调控
实际上,对自旋交叉化合物来说温度调控是最普遍的.通常要确定一个化合物是否具有自旋交叉性质、是否自旋可控,首先要做的就是测定其变温磁化率和(或)变温结构数据,这样就可以直接进行判断.绝大多数化合物的自旋交叉行为都是受温度控制的,在此基础上才有所谓的光激发、压力诱导等,而且不是所有的自旋交叉化合物都具有光和压力调控性质.当然,反过来也有部分化合物在正常温度范围内没有自旋交叉性质,但也可以获得光激发或压力诱导的自旋态转变.
3.5 光学调控
以化合物1的晶型III为例(图13),该晶型两种金属中心的自旋态可以利用温度分别控制,金属中心FeA在室温时为高自旋,在200 K时可转变为低自旋;而金属中心FeB在200 K时为高自旋,在150 K时可转变为低自旋.在极低温时,利用532 nm的激光将两种金属中心的自旋态都激发到高自旋态,在热驰豫的过程中也出现了两步的高自旋到低自旋转变,这与非光激发状态下的温度诱导正好反向同步.
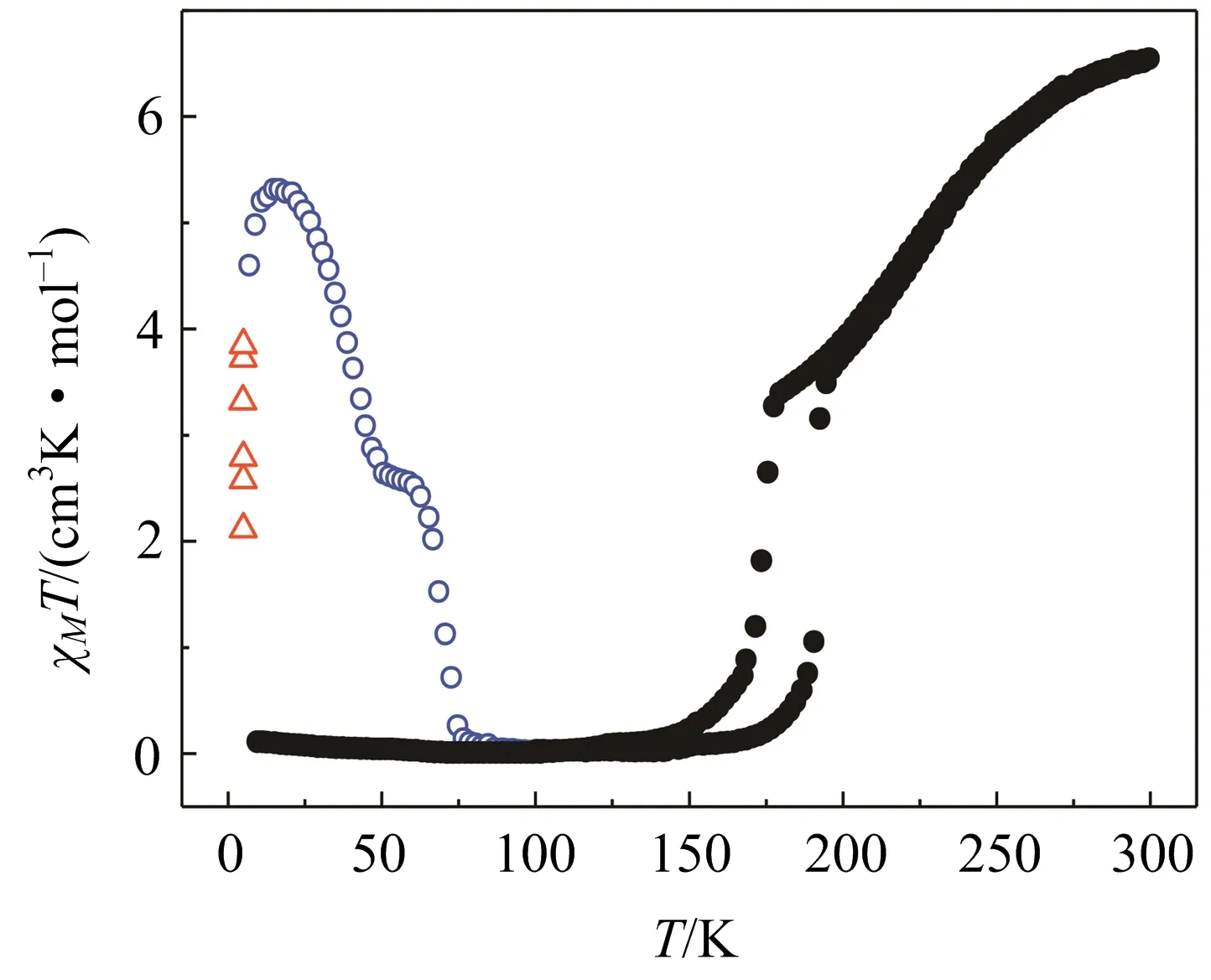
图13 温度对化合物1 晶型III的自旋调控
3.6 力学调控
如前所述,施加压力可以使自旋交叉化合物的金属自旋态从高自旋向低自旋转变.以化合物1为例,在外压条件下一步的自旋交叉行为转变为两步,且两步的转变温度都向高温区移动,也就意味着低自旋态越来越稳定(图14).化合物1的甲醇化晶体本来是两步的自旋转变,在外压下逐渐转变成一步自旋转变,且在较高压条件下基本处于低自旋态.虽然从理论上∂T/∂P= ΔV/ΔSH→L看不到其他因素的影响,实际上压力对自旋态的调控还受晶格稳定性、溶剂分子以及分子间作用力等因素的影响.

图14 压力对化合物1 的自旋调控
笔者课题组利用力学方法对一系列自旋交叉化合物的电子自旋进行了调控,并把这个方法扩展到了其他的双稳态分子体系.如化合物[{Co(dpqa)}2(dhbq)]·(PF6)3(3, dpqa = di(2-pyridylmethyl)-N-quinolin-2-ylmethyl)amine, dhbq = deprotonated 2,5-dihydroxy-1,4-benzoquinone,图15) 在温度变化时可在低温态LS-CoIII-dhbq3-和高温态HS-CoII-dhbq2-之间相互转变,说明存在温度诱导的金属离子与有机配体的电子转移.实际上这个过程中间还存在一个瞬间的过渡态LS-CoII-dhbq2-,也就是说还存在一个CoII离子的自旋交叉过程.当给这个化合物的晶体施加一个外压时,其磁性变化说明金属与有机配体之间的电子转移过程被抑制,只发生了CoII离子的自旋交叉,而且其自旋转变完成率增高[22].
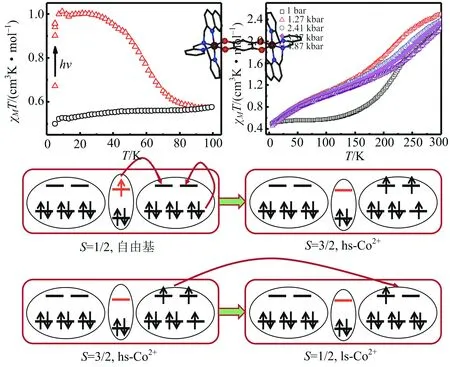
图15 化合物3 自旋态的光学和力学调控
4 自旋交叉-荧光双功能材料
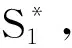
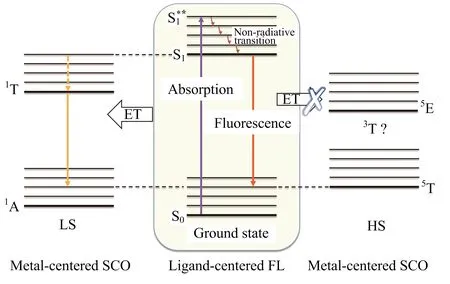
图16 自旋交叉与荧光之间的关联机制
目前自旋交叉与荧光的结合方式主要有两种:物理混合和化学融合.物理混合是指将自旋交叉化合物和发光化合物直接混合在一起,或制成类似于纳米核壳结构的材料;化学融合则是通过化学键将自旋交叉活性中心和发光基团合成在一个分子或体系中,由于自旋交叉活性中心与发光基团通过化学键直接作用,自旋转变与荧光的耦合效果会比物理混合强.物理混合有利于保留自旋交叉化合物的性质,而化学融合则有可能使自旋交叉活性中心散失自旋转变的性质.
笔者课题组在自旋交叉-荧光双功能材料方面的研究起步于2015年,所采取的结合方法介于物理混合和化学融合之间.首先合成了一维自旋交叉化合物[Fe(NH2tz)3]·(ClO4)2(4, NH2tz = 4-amino-1,2,4-triazole)的纳米颗粒,然后不是将荧光基团直接与之混合,而是利用一维结构上的氨基将荧光基团通过氨醛缩合或氨羧缩合反应后修饰到纳米颗粒表面[24],分别获得了2个发光的自旋交叉双功能材料4·芘缩醛和4·罗丹明B(图17),发现在修饰后一维链的自旋交叉性质得以保持,只是转变温度发生较小的变化.变温荧光光谱测试表明发色团的荧光强度随温度和金属自旋态的变化呈现奇特的性质,与预测的结果并不相同.发色团的荧光在低温时并没有淬灭,而是最强的,似乎表明发色团和低自旋金属中心之间并没有发生能量转移,而且随着温度的逐渐升高,荧光强度逐渐减弱,也预示荧光强度只受热效应影响.但是,当温度升高到Fe(II)自旋转变的临界温度Tc时(从∂(χMT)/∂T获得),发色团的荧光强度突然增强,温度继续升高后荧光强度又回到热淬灭过程.这种现象表明荧光强度确实是受到温度变化主导的,但又与自旋转变发生关联,说明两者之间又是有能量转移的.

图17 自旋交叉-荧光双功能材料
笔者课题组的研究表明,自旋交叉与荧光的关联性并不是与理论预测完全一致的,后续工作表明实际上在自旋交叉-荧光双功能体系中,自旋态转变和荧光之间存在多种形式的关联[25],有理论预测的荧光强度与高自旋态比例一致的(自旋态主导),有荧光强度在自旋转变临界点增强的(温度主导),还有多步自旋转变过程中温度和自旋态同时主导的.因此,要阐释清楚自旋交叉与荧光的关联,还有更多的工作待做.
5 结束语
笔者在双稳态分子的自旋调控与功能化研究过程中开展了大量、系统的工作,该文重点综述了其在其中一个方向—自旋交叉化合物的工作(图18):通过配体设计、溶剂分子的可逆交换、晶型控制以及后期的组装等化学方法对基于一个单核分子为母体的一系列化合物的自旋进行了调控,同时还研究了温度、光学和力学等物理方法对这些化合物自旋态的调控.在此基础上,根据Fe(II)离子激发态与有机荧光基团激发态之间的对称性和能级的关联性,采用后合成方法成功地将自旋态转变和荧光变化关联起来,获得了自旋交叉-荧光双功能材料.
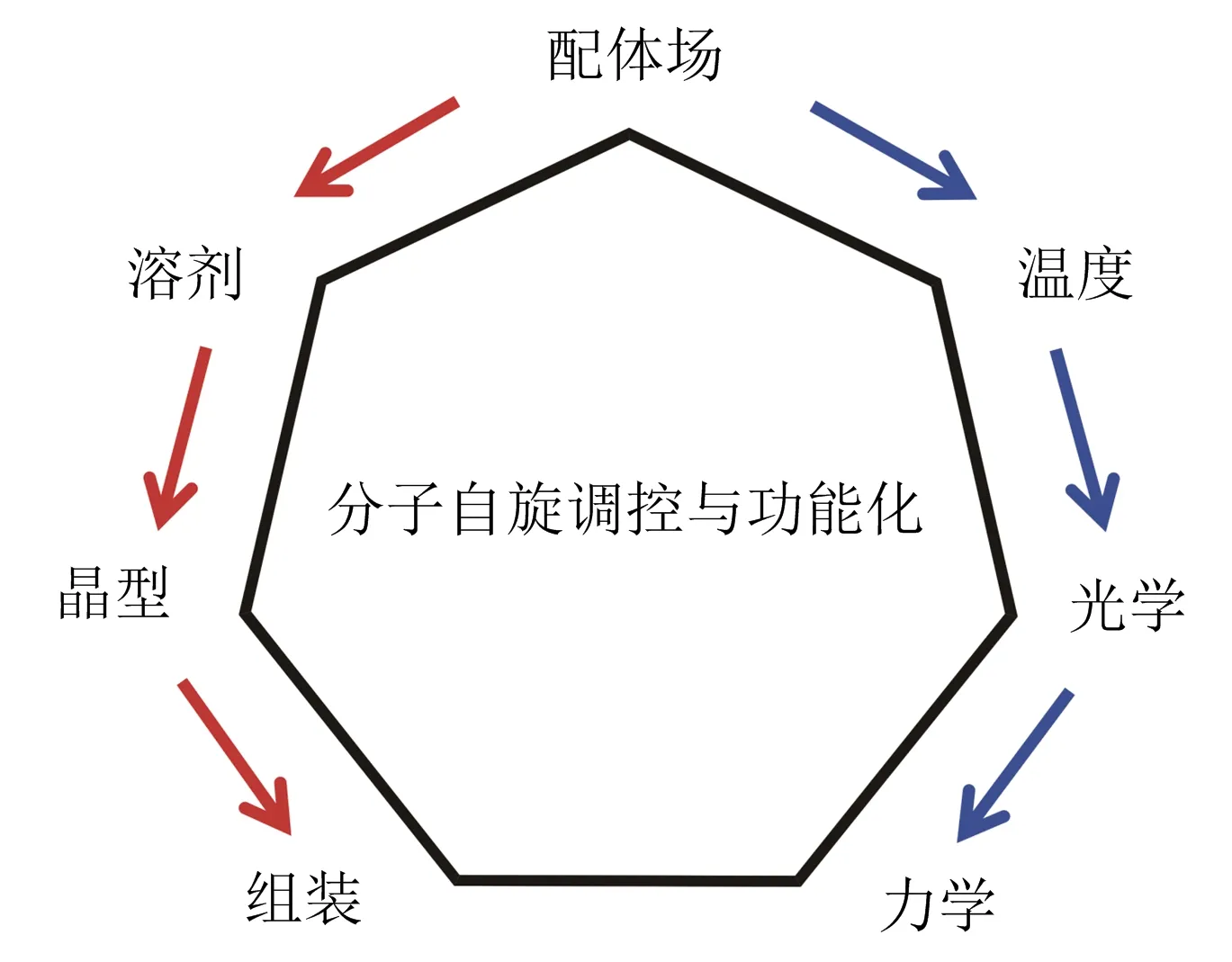
图18 自旋交叉分子的自旋调控示意图
