青海地区藏族HIF-2α基因单核苷酸多态性与胃癌易感性※
2018-09-28马金华王丽娟李进章马金兰骆玉霜沈存芳马金祥徐晓宁
马金华,王丽娟,李进章,马金兰,骆玉霜,沈存芳,马金祥,李 豪,徐晓宁
(1.青海大学附属医院肿瘤内科,青海 西宁 810001;2.青海大学附属医院肾病内科,青海 西宁 810001;3.青海大学医学院,青海 西宁 810001)
青海地区位于青藏高原,胃癌属该地高发肿瘤[1-2]。缺氧诱导因子(hypoxia-inducible factors,HIFs)是参与缺氧应答的转录因子家族之一,可以对多种基因的表达进行调控,并介导某些病理过程[3-4]。现有研究证实HIF-1SNP与胃癌发病相关,HIF-2α单核苷酸多态性(single nucleotide polymorphism,SNP)与胃癌是否相关未见报道。本研究在基因水平检测藏族健康者及胃癌患者血液HIF-2αSNP,分析两者在胃癌发生发展中可能存在的相关性。
1 资料与方法
1.1 组织标本
在青海大学附属医院、青海省人民医院、青海省藏医院,收集2014年11月~2016年12月体检的世居青海的藏族健康者外周血62例作为对照组、45例胃癌患者外周血作为胃癌组。胃癌组男性29例,女性16例;年龄为20~79岁,平均年龄为(48.26±7.32)岁;所有患者病理分型皆为腺癌。对照组男性38例,女性24例;年龄为22~83岁,平均年龄为(46.45±6.51)岁;所有患者病理分型皆为腺癌。一般资料具有可比性(P<0.05)。本课题经过青海大学附属医院医院伦理委员会批准。
1.2 主要仪器与试剂
1.2.1 仪器
PCR扩增仪(Verity 96well,美国 ABI),凝胶成像仪(Gene Genius,英国Syngene),3730XL测序仪(美国 ABI)。
1.2.2 试剂
DNA marker(加拿大 BBI公司),基因组DNA提取试剂盒、PCR扩增试剂盒(上海生工生物工程有限公司)
1.3 实验方法
1.3.1 血样标本收集
空腹静脉采集胃癌组和对照组成员外周血,作EDTA抗凝处理。
1.3.2 基因组DNA提取
取上述血液样品,严格按照上海生工生物工程有限公司的血液基因组DNA提取试剂盒要求提取DNA,用0.8%的琼脂糖凝胶电泳检测完整度,用NanoDrop 2000分光光度计测其含量及纯度后保存于-20 ℃冰箱备用。
1.3.3 引物设计和合成
通过美国国家生物信息中心(NCBI)的GenBanb获取HIF-2α基因序列及其相应位点的SNP信息。并采用PrimerExpress 2.0软件(美国Applied Biosystem Ine.)辅助设计引物。所有引物由上海生工生物有限公司合成,引物序列及扩增片段大小见表 1。
1.3.4 PCR扩增
以基因组DNA为模板,将HIF-2α rs13419896、rs7598371、rs6715787的引物分别建立25 μL PCR反应体系:DNA模板1 μL,上下游引物各0.5 μL,dNTP 0.5 μL,Taq buffer 2.5 μL,Taq酶(5U/μL)0.2 μL,ddH2O 14.8 μL。PCR反应程序:预变性95 ℃ 3 min,变性94 ℃ 30 s,退火58 ℃ 30 s,延伸72 ℃ 45 s,共35个循环;72 ℃ 10 min。上述反应结束后取5 μL PCR扩增产物上样,在1%琼脂糖凝胶上电泳(100V)40 min,用溴化乙锭染色后通过凝胶成像系统照相并保存。扩增产物经纯化后送上海生工生物有限公司测序并进行 SNP 技术分型,根据测序结果确定基因型和基因频率。
表1HIF-2αrs13419896、rs7598371、rs6715787SNP位点引物序列及扩增片段大小

Table 1The order of sequence and the size of amplification fragment
1.4 统计学方法
对照组基因型分布采用Hardy-Weinberg遗传平衡定律检验。数据分析采用SPSS 22.0软件,各基因型分布差异采用χ2检验,α=0.05。
2 结果
2.1 基因组DNA电泳检测结果
胃癌组和对照组血液做基因组DNA提取后,行DNA琼脂糖凝胶电泳检测,并通过凝胶成像仪成像,结果见图1。

1-8:对照组,9-16:胃癌组,M:marker
图1基因组DNA琼脂糖凝胶电泳图
Figure1TheagaroseelectrophoresispictureofgenomeDNA
图1示,基因组DNA提取成功,条带清晰,可以进行后续实验。
2.2 PCR产物检测结果
以2.1所述基因组DNA为模板,进行PCR扩增后,再经1%琼脂糖凝胶电泳,结果见图2~4。
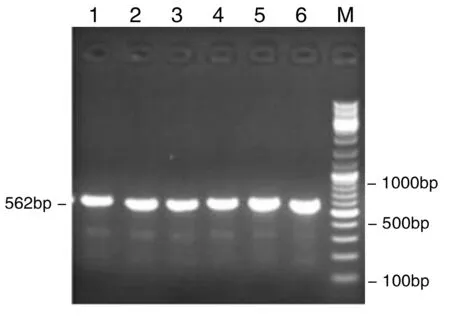
图2HIF-2αrs13419896基因PCR扩增图
Figure2TheHIF-2αrs13419896genePCRamplification
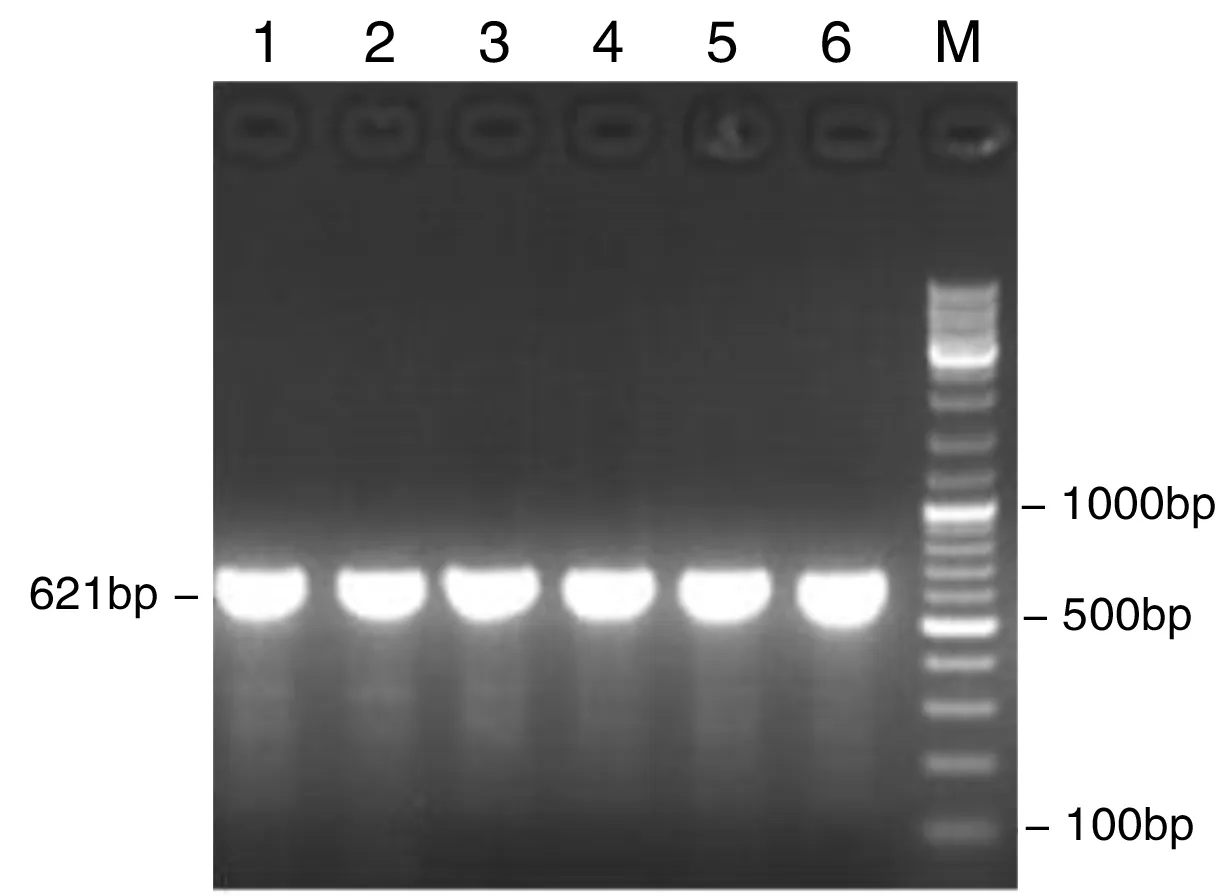
图3 HIF-2α rs7598371基因PCR扩增图

1-3:对照组,4-6:胃癌组,M:marker
图2~4示,HIF-2α rs13419896、rs7598371和rs6715787位点扩增片段大小分别在562 bp、621 bp和513 bp处。
2.3 测序结果
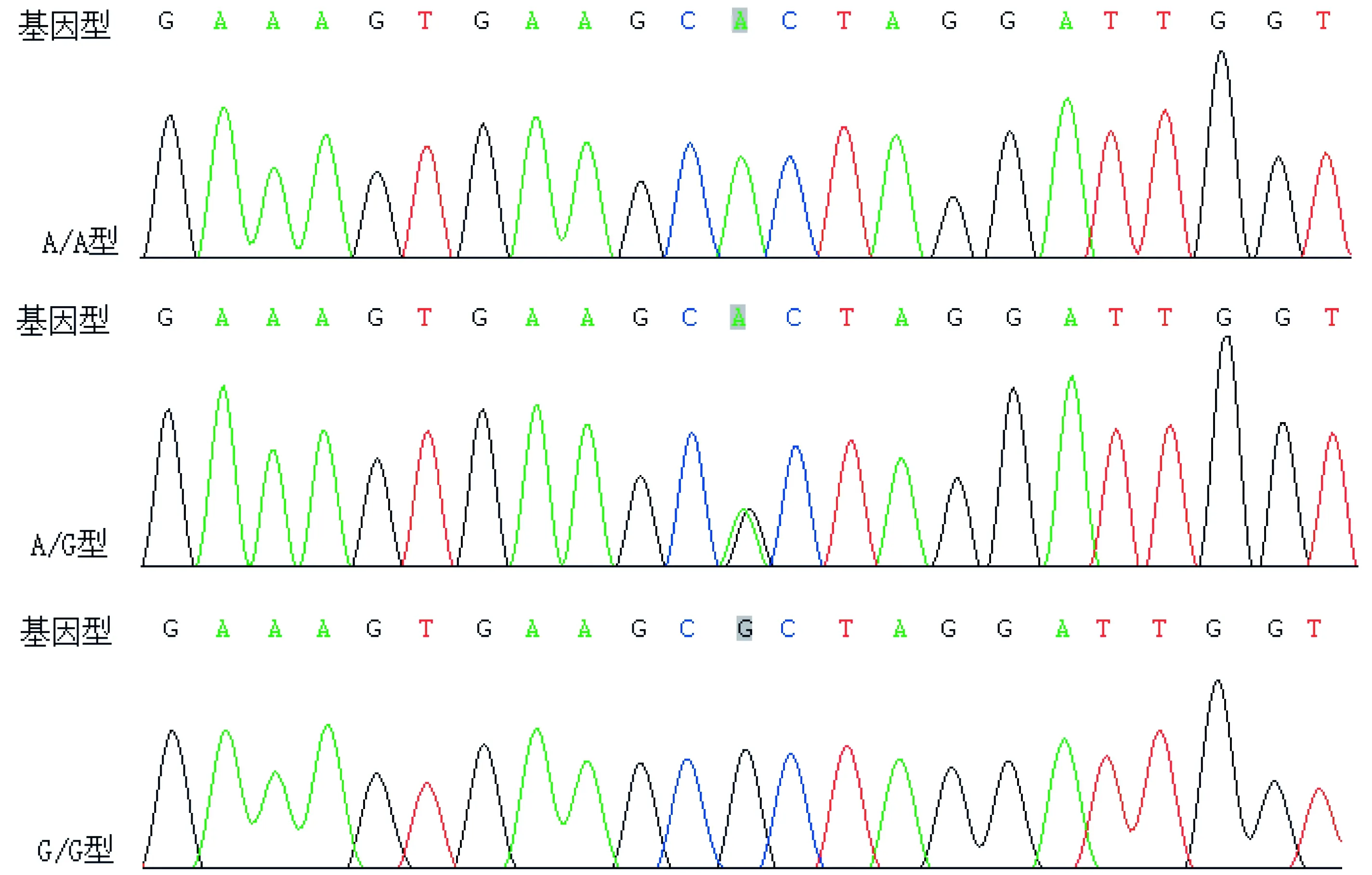
图5HIF-2αrs13419896SNP基因型图
Figure5ThepictureofHIF-2αrs13419896SNPgenotype
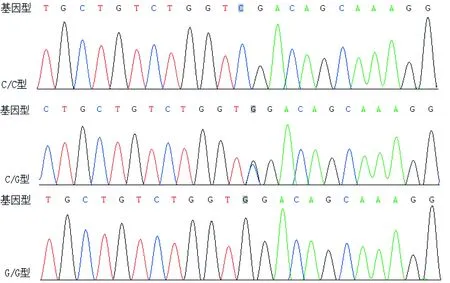
图6 HIF-2α rs7598371 SNP基因型图


图7 HIF-2α rs6715787 SNP基因型图
图5~7示,HIF-2α rs13419896 SNP基因型分别为A/A、A/G、G/G型,rs7598371 SNP基因型分别为C/C、C/G、G/G型,rs6715787 SNP基因型分别为C/G、C/C、G/G型。
2.4 基因型和基因频率在两组中的分布结果
检测结果示,rs13419896SNP基因型分别为A/G、G/G、A/A,rs7598371SNP基因型分别为C/G、C/C、G/G,rs6715787SNP基因型分别为C/G、C/C、G/G。对照组检验基因型分布符合Hardy-Weinberg平衡规律(rs13419896SNP:χ2=3.07,P>0.05;rs7598371SNP:χ2=1.54,P>0.05;rs6715787SNP:χ2=2.28,P>0.05)。rs13419896的A/G、G/G、A/A基因型频率在胃癌组中分别为28.89%、28.89%、42.22%,在对照组中的频率分别为38.71%、27.42%、33.87%。rs7598371的C/G、C/C、G/G基因型频率在胃癌组分别为26.67%、35.56%、37.78%,在对照组分别为41.94%、25.81%、32.26%。rs6715787的C/G、C/C、G/G基因型频率在胃癌组分别为35.56%、40.00%、24.44%,在对照组分别为40.32%,32.26%、27.42%。其中rs13419896位点的基因型分布频率在胃癌组中A/A型最高,在对照组中A/G型最高。rs7598371位点的基因型分布频率在胃癌组中G/G型最高,在对照组中C/G型最高。rs6715787位点的基因型分布频率在胃癌组中C/C型最高,在对照组中C/G型最高,经χ2检验示,胃癌组与对照组间均无统计学差异(P>0.05),具体数值见表2~4。
表2rs13149896基因型和等位基因频率分析结果表(%)

Table 2The analysis of rs13149896 genotype and allele frequency(%)
表3rs7598371基因型和等位基因频率分析结果表(%)

Table 3The analysis of rs7598371 genotype and allele frequency(%)
表4rs6715787基因型和等位基因频率分析结果表(%)

Table 4The analysis of rs6715787 genotype and allele frequency(%)
3 讨论
本研究结果显示,青海地区藏族HIF-2α基因单核苷酸多态性与藏族胃癌易感性无关。
胃癌是青海地区高发肿瘤。有学者提出,各地区、各民族胃癌发生率不同的原因是胃癌的发生为多重基因、多种因素共同作用的结果[5]。临床上对HIF-1α与HIF-2α的研究比较多[6]。其中,HIF-1是调节亚基,它于1992年被Semenza等人发现。其可调控VEGF的表达,以此控制肿瘤的发生、发展[7]。HIF-2α是HIF-1的功能亚基,其可识别并结合调控基因中的缺氧反应元件(hypoxia response element,HRE)调控下游基因合成,以此调节肿瘤血管生成、浸润和远处转移等[8-10]。HIF-2α是参与缺氧调节的关键转录因子之一,在生物体的正常生理调节和肿瘤发生、发展过程中均发挥重要作用[11,12]。HIF-2α与胃癌的关系同样十分密切,2010年Wang等人研究发现HIF-2α在胃癌中的作用类似HIF-1α,提示 HIF-2α在胃癌的病理阶段和转移中发挥作用[13]。因此,HIF是肿瘤发生、发展和治疗研究的热点[14]。
单核苷酸多态性(SNP)是由基因组核苷酸水平上的变异引起的,绝大多数SNPs本身虽不是易感性的原因,但在全基因组范围内比较易感和非易感人群之间的SNP图谱,可显示易感人群基因组的结构特点并通过关联分析寻找易感基因。
研究青海地区藏族HIF-2α基因rs13419896、rs7598371、rs6715787位点的多态性以及它们与胃癌发生、发展的相关性具有重要意义。若能明确它们的相关性,便可以为高危人群提供预警信号[15]。本研究采用的主要研究技术为“巢式PCR-RFLP”基因分型技术,这种技术相比较于传统的SNP分型技术来讲,可以对任何位点行常规性限制内切酶鉴定,同时该SNP分型技术利用巢式PCR技术,使得对多个位点同时进行分析成为可能。而SNP分型技术只能对有酶切位点的部位进行切割,这就在一定程度上可以实现对多个位点进行分析。因此,即便所得的DNA浓度较低,也可以对其进行快速的分型[16]。青藏高原为低氧环境,患者体内的HIF-2α容易被过度激活,导致患者体内的红细胞明显增多及组织血管生成紊乱,进一步导致高原病的发生。而长期筛选形成的基因多态性对于藏族人群来讲,可以使其适应高原的环境,在低氧环境下亦能满足自身的需求。有学者认为基因多态性是对种族的正选择。我国和日本、英国、加拿大、美国以及尼日利亚科学家共同合作创建了HapMap项目,此项目的主要目的是为了确定人类遗传基因的相似性以及差异性,以此来揭示多态信息的单体型图,探究遗传基因的奥秘。现阶段,HapMap标签单核苷酸分布(SNPs)数据已然成为了科研中的参考数据之一。在实验中,对目的基因进行扩增检测,并将其信息与HapMap数据库中的信息进行比对,可发现个体差异基因与疾病之间的关系[17]。
肿瘤细胞与正常细胞的相同点是,在生长过程中皆会出现局部的缺血,进而导致氧气供应不足。HIFα家族转录因子是生物体对缺氧条件做出应答的关键性调节因子。在肿瘤中,HIF蛋白会因VHL基因的突变使其失调,这一基因的突变会阻碍VHL通过其泛素连接酶活性对HIF蛋白起负向调节作用。这一观点已经在HIF-2α中得到了证实,HIF-2α是HIF的一种异构体形式,这种形式在VHL突变型肿瘤中很容易发生上调,最近有报道称其是肿瘤干细胞的驱动因素。然而,我们还不清楚有哪些信号传导作用将干细胞的内在转录作用机制与肿瘤中HIF-2α调节作用机制联系在一起[18]。
HIF-1α的表达和活性的调节是有氧浓度依耐性的,氧诱导的羟化酶的作用途径是调节HIF-1最重要的途径。
HIF-1α转录调节因子,特别是广为人知的HIF-2α(亦称EPAS1),会在肿瘤干细胞中表达而且是维护肿瘤干细胞状态所必需的。然而ID2在CSCs中通过哪些途径促进HIF-2α在肿瘤干细胞中积累我们并不清楚。在此我们报道DYRK1A和DYRK1B激酶会对ID2上的27号苏氨酸(Thr27)进行磷酸化。缺氧条件会通过使DYRK1A和DYRK1B的失活而使这种磷酸化作用降低。这些激酶的活性会在常氧的条件下通过对氧具有感知作用的脯氨酰羟化酶PHD1(亦称EGLN2)被激活。ID2会与VHL泛素连接酶复合物结合,取代与VHL有相关性的Cullin 2,对HIF-2α的泛素化和降解作用进行破坏。由DYRK1对ID2的Thr27的磷酸化作用会阻断ID2与VHL的相互作用,从而维护HIF-2α的泛素化作用[19]。
综上所述,探究藏族HIF-2α基因位点的多态性以及它们与胃癌的相关性具有积极意义。本研究结果尚待进一步扩大样本量和更进一步的分型人群确认。
