咪唑并[1,5-a]吡啶的合成工艺研究
2018-09-18石加威
姜 坤, 石加威, 刘 溪
(1.淮南联合大学 化学工程系,安徽 淮南 232038;2.安徽理工大学 化学工程学院,安徽 淮南 232001)
咪唑并吡啶类杂环化合物是一种重要的含氮杂环类化合物,因与吲哚、氮杂吲哚等杂环具有较大的结构相似性,可以作为吲哚的生物电子等排体用于药物的结构优化,用于新药的研发[1]。许多咪唑并吡啶类杂环化合物已被用于肿瘤、微生物感染、胃溃疡和失眠等疾病的药物研发。此外,咪唑并吡啶类杂环化合物还具有较好的光电物理化学性能, 它也被广泛应用于光电材料领域[2]。
由于含氮杂环在材料、生物医药领域有着广泛的应用,长久以来,含氮杂环化合物都是有机合成领域的研究热点,化学家们一直致力于开发各种新型的合成方法来高效地构建含氮杂环化合物。传统上,咪唑并[1,5-a]吡啶类杂环化合物的合成主要是利用N-吡啶甲基酰胺作为底物,通过维尔斯迈尔型环化反应得到[3]。2012年采用单质碘为促进剂,当量的无机碱作为添加剂,中国科学技术大学汪志勇等实现了2-吡啶甲醛与氨基酸的脱羧偶联环化反应,能在室温条件下合成咪唑并[1,5-a]吡啶[4]。随后,南阳师范学院徐坤等采用铜盐为催化剂,2-苯甲酰基吡啶分别和苯甲胺[5]或氨基酸反应[6],成功合成了一系列咪唑并[1,5-a]吡啶。然而,以上方法均需要预先制备含官能团的底物(醛或酮),限制了以上方法的适用范围。因此,开发新型的合成方法用于高效的构建咪唑并[1,5-a]吡啶类杂环化合物具有重要意义。利用过渡金属催化碳氢键的直接胺化策略,可以避免反应底物的预先官能团化,已经成为形成碳氮键的重要途径。本文采用了一种基于碳氢键断裂的碳氮键形成策略,用来合成咪唑并[1,5-a]吡啶类杂环化合物的方法,该方法利用过渡金属催化的多步串联反应,可以从便宜易得的底物一锅法制备咪唑并[1,5-a]吡啶类杂环化合物。
1 材料与方法
1.1 仪器与试剂
仪器:气相色谱-质谱联用(岛津QP2010),三用紫外分析仪(ZF-1),薄层硅胶板(GF254),Avance 400MHz型核磁共振仪(瑞士Bruker公司),恒温磁力搅拌器(德国IKA),分析天平(梅特勒-托利多),旋转蒸发仪(上海亚荣生化仪器厂)等。
试剂:2-苄基吡啶、苯甲胺、溴化亚铜二甲硫醚、特戊酸、甲苯、1,2-二氯乙烷(DCE)、N,N-二甲基甲酰胺(DMF)、二甲基亚砜(DMSO)、石油醚、乙酸乙酯、无水硫酸钠、柱层析硅胶(200~300目)等为分析纯,均采购于国药集团化学试剂有限公司;GF254预制薄层层析板购于青岛海洋化工有限公司。
1.2 咪唑并[1,5-a]吡啶的合成
取一支干燥的两口反应管,加入磁力搅拌子,依次向反应管中加入溴化亚铜二甲硫醚(0.06 mmol)、特戊酸(0.3 mmol),再将2 mL甲苯加入反应管,室温搅拌5 min;通过微量注射器分别将苄基吡啶(0.3 mmol)和苄胺(0.6 mmol)加入到上述混合液中,室温搅拌5 min后,接上冷凝回流管;然后将反应管置入油浴中,升温到110 ℃;随后,再加入苄胺(0.3 mmol)继续反应约12 h。用薄层层析监测反应至中间体2-苄基吡啶消失,停止反应。将反应液冷却至室温,再把反应液转移到分液漏斗中,加入30 mL水,用乙酸乙酯为萃取剂进行萃取操作(3×10 mL);萃取结束后,用无水硫酸钠干燥乙酸乙酯层;旋干乙酸乙酯溶液,用200~300目硅胶作为固定相,石油醚乙酸酯的混合液作为洗脱剂,柱层析分离得到咪唑并[1,5-a]吡啶。
1.3 反应条件优化
本文选择苄基吡啶和苄胺为模型底物,采用单因素试验法,对催化剂、添加剂、溶剂、反应温度、反应时间和投料比等条件进行优化,以期获得较好的反应条件。
1.4 反应的适应性研究
在获得优选的反应条件基础上,通过改变底物的取代基,以考察该方法的适用范围。
1.5 目标化合物的结构解析
综合利用核磁共振氢谱、碳谱来分析目标产物的结构。
2 结果与分析
2.1 反应条件优化
对催化剂、添加剂、溶剂、反应温度、反应时间和投料比等条件进行优化,以期获得较好的反应条件。化学反应式如图1所示。
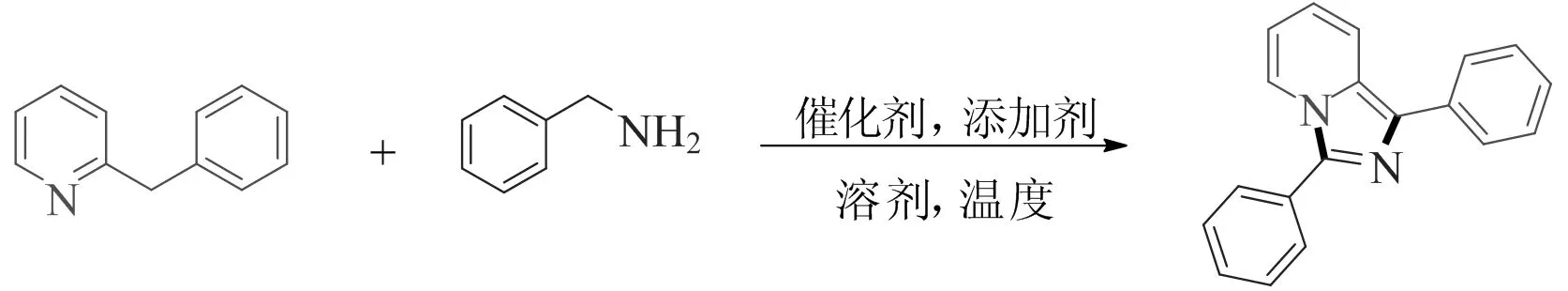
图1 化学反应式
2.1.1 催化剂的优化 如表1所示,首先尝试了醋酸铜作为催化剂,对甲基苯磺酸作为添加剂,甲苯作为溶剂,温度控制在110 ℃,反应24 h,能以47%的分离产率得到目标产物(Entry 7)。将醋酸铜换为醋酸钴或醋酸锰,都没有检测到目标产物的生成(Entry 2~3),说明铜盐对该反应起着至关重要的作用。于是,考察别的铜盐对反应的影响。当碳酸铜、碘化铜、三氟甲磺酸铜、溴化亚铜二甲硫醚加入到反应体系作为催化剂,均可以较好的收率得到目标产物(Entry 4~7)。而不加催化剂时,也没有检测到目标产物生成(Entry 8)。比较试验数据可知,当溴化亚铜二甲硫醚作为催化剂时,得到的目标产物的收率是相对最高的(Entry 7)。因此,选择溴化亚铜二甲硫醚作为该反应的催化剂。
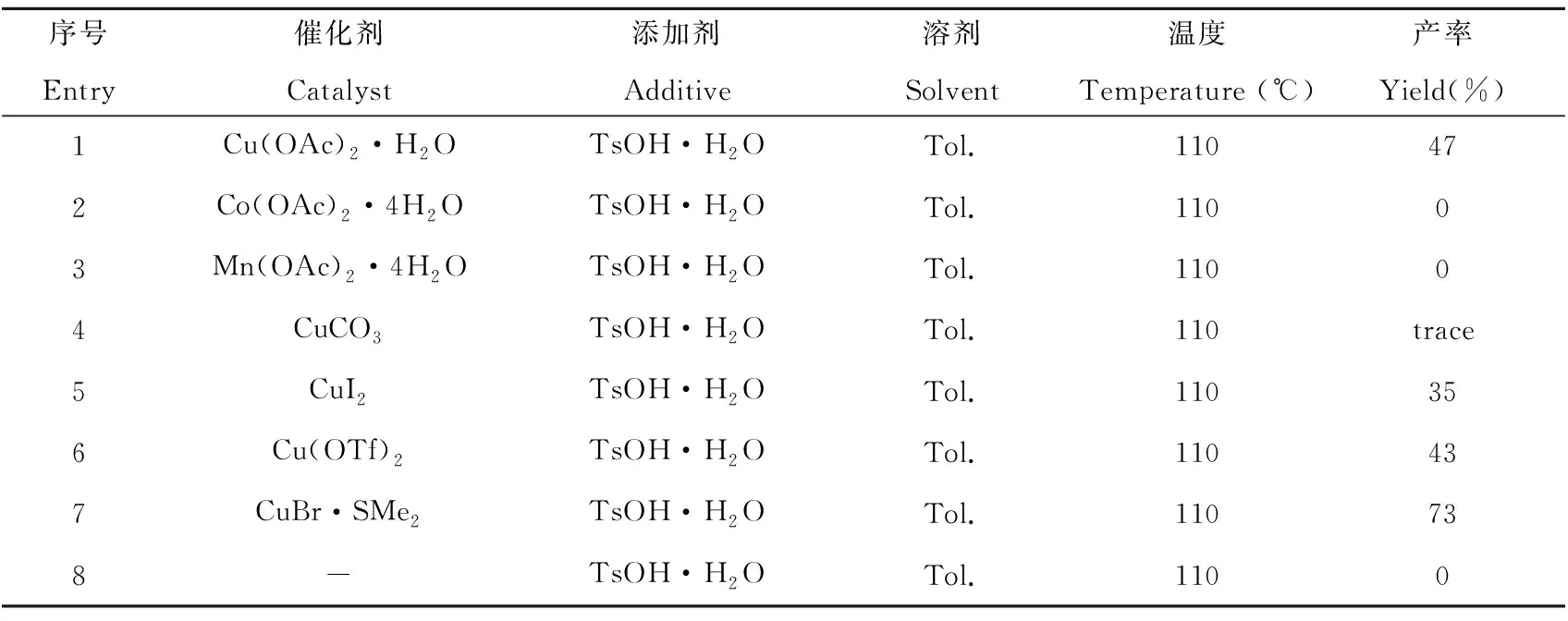
表1 催化剂优化
2.1.2 添加剂的优化 在反应底物的种类与用量完全相同的情况下,选择溴化亚铜二甲硫醚作为催化剂,甲苯作为溶剂,温度控制在110 ℃,将对甲苯磺酸替换为别的添加剂,考察添加剂对反应的影响。当采用质子酸,如特戊酸、乙酸、苯甲酸、三氟乙酸时,均能以较好的收率得到目标产物(Entry 1~5)。当体系中存在三氟乙酸时,采用GC-MS,可以检测到有大量三氟乙酰苯甲胺产生,导致产率相对较低。为了避免生成酰胺副产物,于是选用具有较大空间位阻的特戊酸作为添加剂,可以86%的分离产率得到目标产物(Entry 5)。选用路易斯酸(三氟化硼乙醚)作为添加剂也能很好的促进反应,以50%的分离产率得到目标产物(Entry 6)。随后,我们还尝试了用无机碱作为添加剂进行试验,发现反应中几乎没有目标产物生成(Entry 7~8)。另外,在不加入添加剂的情况下(Entry 9),几乎没有目标产物生成。综合考虑,特戊酸是相对比较适合该类反应的添加剂。因此,最终我们选择特戊酸作为该反应的添加剂。
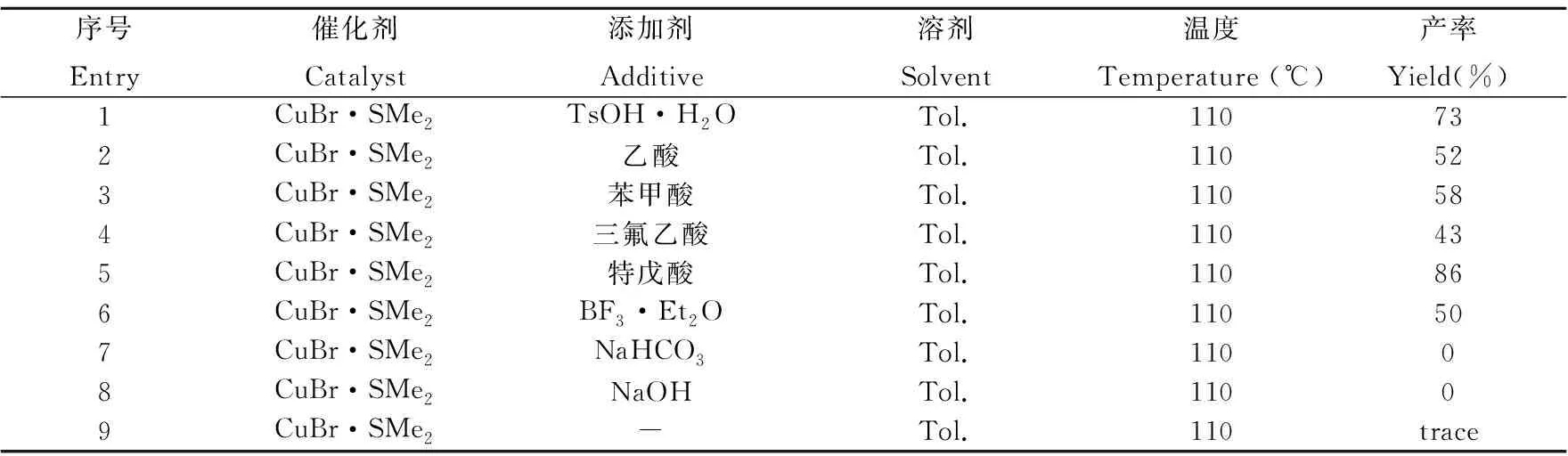
表2 添加剂的优化
2.1.3 溶剂的优化 在反应底物的种类与用量完全相同的情况下,将优选的溴化亚铜二甲硫醚作为催化剂,特戊酸作为添加剂,温度控制在110 ℃,然后考察溶剂对反应的影响。由表3可知,当把溶剂由甲苯换为1,2-二氯乙烷、1,4-二氧六环、硝基甲烷或水作为溶剂时,几乎没有目标产物生成。当选用N,N-二甲基甲酰胺、二甲基亚砜、正丁醇、三氟甲基苯为溶剂时,产率大幅下降。而选用氯苯为溶剂时,可以53%的分离产率得到目标产物(Entry 6)。由此可知,当甲苯作为该类反应的溶剂时,获得的收率是相对比较理想的。因此,选择将甲苯作为该反应的溶剂。
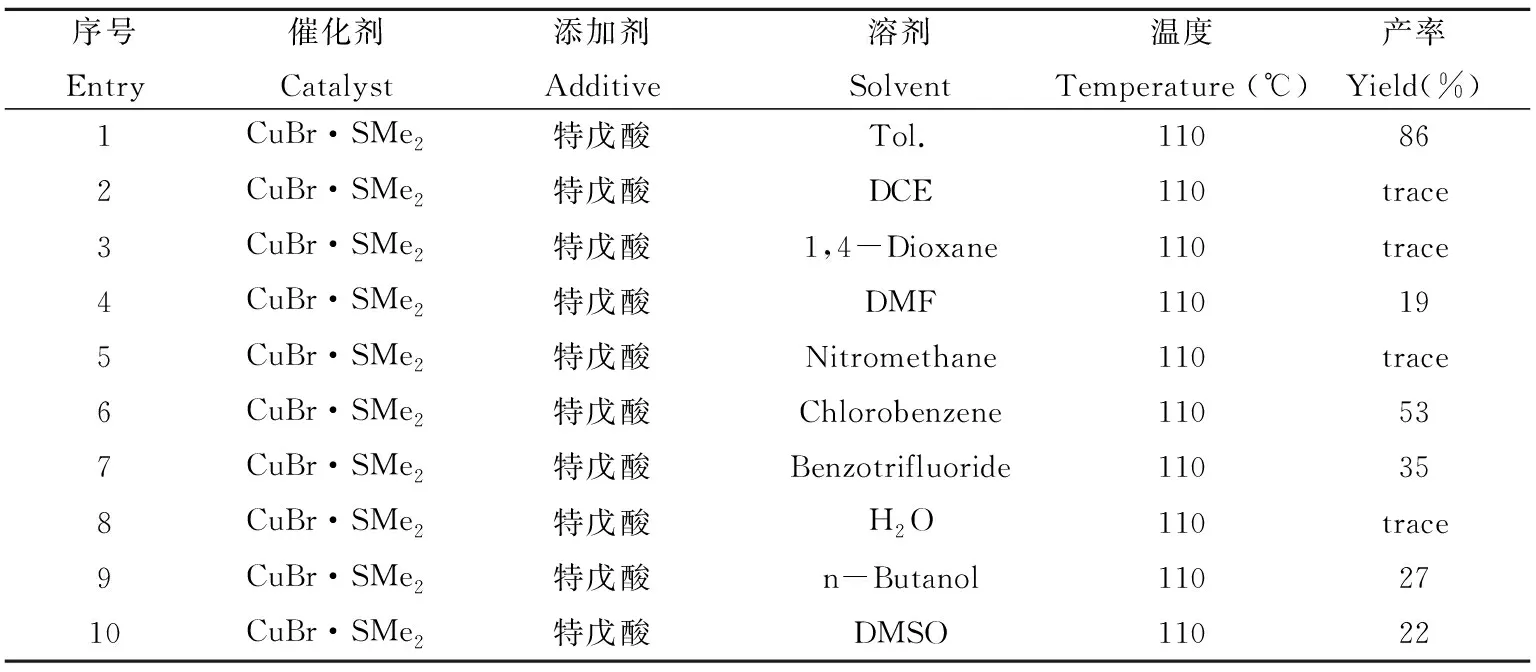
表3 溶剂的优化
2.1.4 其它反应条件的优化 在以上优化试验的基础上,考察了温度对反应的影响。当将温度由110 ℃升高为120 ℃进行反应时,反应体系变得较乱,杂质点明显增多,以61%的分离产率得到目标化合物;随后尝试降低反应温度到100 ℃,反应24 h后,发现有许多苄基吡啶不能转化,以46%的分离产率得到目标产物。所以,选择110 ℃为本反应的反应温度。
在前期的优化实验中,我们发现有许多2-苯甲酰基吡啶产生,这些中间体在24 h内不能完全转化为产物。于是,尝试将苄基吡啶和苄胺的投料比由1∶2升高为1∶3。试图通过增加苄胺的量,让苄胺与2-苯甲酰基吡啶反应,来生成目标产物。然而,效果并不理想,依然能监测到中间体的存在。考虑到苄胺稳定性差,且苄基吡啶转化为中间体2-苯甲酰基吡啶也需要一定时间,故调整了一下苄胺的投料方式。一开始是两当量的苄胺和一当量的苄基吡啶一起反应12 h,随后再补加一当量的苄胺,继续反应12 h,最终以92%的分离产率得到目标产物1,3-二苯基咪唑并[1,5-a]吡啶。
2.2 反应的适应性研究
在获得优选的反应条件基础上,通过改变底物的取代基,以考察该方法的适用范围(图3)。苯甲胺的苯环上引入给电子的甲氧基,产率有小幅降低(3b)。而苯环上引入吸电子的三氟甲基,则目标产物(3c)的产率上升到94%。含有卤素取代基也能很好的兼容此反应,能以较好的收率获得目标产物(3d~3f),这为进一步借助偶联反应来对目标分子进行结构修饰提供了可能。从图3可知,目标产物(3d)的产率小于目标产物(3f)产率,说明该反应存在一定的位阻效应,导致目标产物(3d)收率降低。
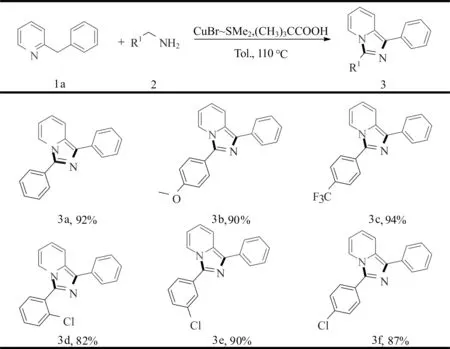
图2 反应的适应性研究
2.3 目标化合物的结构解析
本文综合利用核磁共振氢谱、碳谱来确定目标产物的结构。谱图解析结果表明试验测得数据与目标化合物的结构相匹配。代表性目标产物结构表征数据如下:
(3a)1H NMR (400 MHz, CDCl3) 8.22-8.20 (d, J=7.2 Hz, 1H), 7.95 (s, 1H), 7.93 (s, 1H), 7.83-7.81 (m, 3H), 7.54-7.41 (m, 5H), 7.31-7.27 (t, J=7.2 Hz, 1H), 6.78-6.74 (m, 1H), 6.56-6.52(t, J=7.2 Hz, 1H).13C NMR (100 MHz, CDCl3) 136.1, 135.0, 132.0, 130.2, 129.0, 128.8, 128.7, 128.3, 127.7, 126.8, 126.5, 121.7, 119.7, 119.1。
(3b)1H NMR (400 MHz, CDCl3) δ8.16 (d, J=7.0 Hz, 1H), 7.94 (d, J=7.5 Hz, 2H), 7.82 (d, J=9.3 Hz, 1H), 7.76 (d, J=8.4 Hz, 2H), 7.47 (t, J=7.5 Hz, 2H), 7.30 (t, J=7.3 Hz, 1H), 7.06 (d, J=8.4 Hz, 2H), 6.86—6.70 (m, 1H), 6.55 (t, J=6.5 Hz, 1H), 3.88 (s, 3H).13C NMR (100 MHz, CDCl3) δ 160.3, 138.0, 134.8, 131.5, 130.0, 128.8, 127.4, 127.0, 126.6, 122.4, 121.9, 119.7, 119.3, 114.6, 113.3, 55.5。
(3c)1H NMR (400 MHz,CDCl3) δ 8.24 (d, J=7.2 Hz, 1H), 7.98 (d, J=8.1 Hz, 2H), 7.92 (d, J=7.2 Hz, 2H), 7.86 (d, J=9.3 Hz, 1H), 7.78 (d, J=8.2 Hz, 2H),7.47 (t, J=7.7 Hz, 2H), 7.31 (t, J=7.4 Hz, 1H), 6.83 (dd, J=9.1, 6.4 Hz, 1H), 6.63 (t, J=6.8 Hz, 1H).13C NMR (100 MHz, CDCl3) δ136.6, 134.7, 133.8, 132.9, 130.5, 128.9, 128.42, 128.36, 127.0, 126.1, 124.1 (d, J=272.1 Hz), 121.7, 120.4, 119.5, 114.1。
3 结论
本文报道了一种铜催化的咪唑并[1,5-a]吡啶的合成方法。以溴化亚铜二甲硫醚为催化剂,特戊酸为添加基,甲苯为溶剂,在空气气氛下,2-苄基吡啶和苄胺发生氧化串联反应,以较高的产率制备出1,3-二取代的咪唑并[1,5-a]吡啶类化合物。该方法所用原料价廉易得,不需要对底物进行预官能团化,减少了合成步骤,具有一定的实用性。
