先天性胎毛性多毛症一例
2018-09-14唐金玲汤建萍
唐金玲,汤建萍
临床资料
患儿,男,3岁6月龄。因全身皮肤多毛3年6个月,于2015年10月就诊。患儿出生时全身皮肤覆盖大量毛发,最长约5 cm,呈兽皮样外观;胸腹、背部、面颊及下颌部毛发自出生后逐渐脱落,就诊时已大部分脱落;左上肢毛发较出生时略有减少,毛发长度稍变短;但其他部位毛发无明显脱落或增多。未治疗。患儿生长发育及智力正常,面部形态正常,有龋病但无牙龈增生及牙齿畸形,无色素痣,无其他伴随疾病。患儿为第2胎第2产,其姊体健,父母非近亲结婚,母亲怀孕期间无特殊接触史、用药史。家族中无类似病史。体格检查:一般情况可,视力正常,右侧第2、3下乳牙,左侧第3上乳牙可见龋齿,上中切牙间隙增宽(约1~2 mm),无牙龈增厚;浅表淋巴结未触及增大。皮肤科情况:患儿眉毛粗黑,额部、四肢及臀部皮肤覆盖大量浓密黑色毛发,最长约3.5 cm,胸腹及背部毛发量稍多但较细软,面颊部、下颌部毛发正常;指(趾)甲正常(图1)。辅助检查:睾丸酮37.66 ng/dl(正常值0~43 ng/dl),硫酸脱氢表雄酮40.3 g/dl(19~230 g/dl),垂体泌乳素5.28 ng/ml(1~19 ng/ml),促肾上腺皮质激素14.9 pg/ml(7.2~63.6 pg/ml),促甲状腺激素(TSH)、游离三碘甲状原氨酸(FT3)、游离甲状腺素(FT4)均正常。胸、腰、骶椎骨骼X线检查均无异常,未见脊柱裂。诊断:先天性胎毛性多毛症。治疗:因患儿毛发持续缓慢自然脱落,逐渐减少并无增多趋势,建议额部暴露部位行激光脱毛治疗,其余部位可暂时观察;同时建议行基因检测。家长因经济原因拒绝进一步检查与治疗。随访2年,患儿体毛无明显变化。
讨论
人类体表的毛发分为毳毛、长毛和短毛3种类型。毳毛毛发细软,色淡,如面、颈、躯干及四肢的毛发。长毛和短毛均为终毛,终毛较长、粗且硬,如头发、睫毛、眉毛、胡须、腋毛、阴毛等。全身性多毛症分为获得性和先天性,前者见于内分泌疾病、营养不良、皮肌炎、药物影响等。先天性全身多毛症(congenital generalized hypertrichosis,CGH)包括先天性胎毛性多毛症(congenital hypertrichosis lanuginose,CHL)和先天性全身终毛增多症(congenital generalized hypertrichosis terminalis,CGHT)。CGH 非常罕见,发病率约为1/10亿[1],往往是复杂畸形综合征的皮肤表现[2]。CHL在1970年由Beighton[3]首次报道。多例报道CHL患儿毛发可以随年龄增长逐渐自行减少或脱落[4,5]。有文献报道患儿可无家族史,无内分泌、代谢和遗传调查的异常[5]。但亦有可追溯到多毛症家族史同时并发牙齿异常、幽门狭窄[4]、竖毛肌附着处的弥漫性错构瘤[6]及先天性青光眼[7]的案例。Bondeson等[8]报道缅甸累及一家系4代的CHL,为常染色体显性遗传模式。CGHT是一组终毛持续过度生长的异质性疾病群体[1],是一种常染色体显性遗传的CGH,常伴发牙龈增生和面部特征性畸形[9]。到目前为止已经鉴定的潜在基因突变位点可能包括8q22、17q24.2-q24.3 和 xq24-q27.1[1,9]。Baumeister等[10]曾在1993年报道非综合征性的CGHT,仅表现为皮肤多毛,无牙龈肥大及其他任何器官系统异常,并称其为阿姆布拉斯综合征(Ambras syndrome),但因此称呼无特定含义且含糊不清容易误导而被废弃[1]。
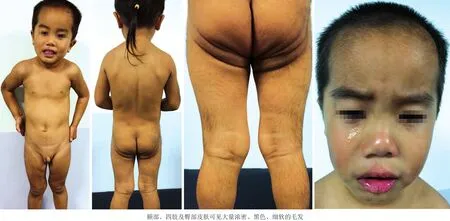
图1 先天性胎毛性多毛症患儿临床表现
目前对于CGH的治疗多为对症治疗,如激光脱毛术等;如并发其他系统病变如牙龈增生、骨骼异常等则给予相应治疗。对于患儿及其家属可行基因检测以发现致病基因。本例患儿出生时全身即有大量毛发,逐渐自然脱落,身体、智力发育正常,辅助检查均正常,据此可诊断为先天性多毛症。由于患儿多毛的部位分别位于额部、胸腹部、背部、臀部及四肢,虽浓密但较细软,与出生时胎毛类似,考虑为胎毛性多毛症,故诊断为CHL。也有学者将CHL翻译为先天性毳毛性多毛症[11],但因毳毛颜色淡甚至无色,与本例患儿的毛发特征不相符,因此笔者认为CHL的中文释义为先天性胎毛性多毛症更为合理。目前该患儿年龄尚小,整体情况尚可,推测其预后较好,但仍需继续观察患儿的生长发育,尤其是内分泌、牙齿及智力情况,有必要长期追踪患儿及其后代的情况。
