彩色棉多药和有毒化合物输出蛋白MATE家族基因的鉴定及表达分析
2018-09-11王作敏孙士超张新宇李艳军
王作敏 刘 瑾 孙士超 张新宇 薛 飞 李艳军,* 孙 杰
彩色棉多药和有毒化合物输出蛋白MATE家族基因的鉴定及表达分析
王作敏1刘 瑾1孙士超2张新宇2薛 飞2李艳军2,*孙 杰2
1石河子大学生命科学学院, 新疆石河子 832003;2石河子大学农学院 / 新疆生产建设兵团绿洲生态农业重点实验室, 新疆石河子 832003
植物多药和有毒化合物输出家族(multidrug and toxic compound extrusion, MATE)是一类可转运阳离子染料、氨基葡糖、多种抗生素与药物等次生代谢产物的转运蛋白家族。本研究利用生物信息学手段从陆地棉基因组数据库中鉴定了MATE家族基因, 并从基因的系统进化关系、染色体分布、基因结构和表达模式等方面对该基因家族特征进行了比较分析。共鉴定出91个陆地棉基因, 命名为~。陆地棉MATE蛋白与拟南芥MATE蛋白均可分为A、B、C、D、E、F和G 7个亚家族, 其中84个GhMATE蛋白具有12个典型的跨膜结构域。染色体定位显示,家族成员定位在不同的25条染色体上, 共形成5个基因簇。qRT-PCR分析发现, GhMATE家族基因在棉花各组织中均有表达, 但表达模式各不相同, 其中和在棕色棉纤维中的表达量明显高于在白色棉中, 表明它们可能与棕色棉纤维的颜色形成相关。本研究为进一步解析棉花MATE家族基因的功能和作用机制积累了有价值的资料。
陆地棉; 彩色棉; MATE家族; 生物信息学; 实时荧光定量PCR
天然彩色棉成熟吐絮时纤维自身具有天然色彩[1]。其纤维在制成成品之前无需化学印染, 既可节省生产成本, 又可避免纺织品中化学染料对人体健康和环境造成的不良影响, 符合当前人们注重健康、环保, 崇尚自然的消费理念, 因此, 彩色棉及其纺织品的开发和利用具有十分广阔的市场前景[2-3]。然而, 天然彩色棉品种资源仅包括棕色和绿色两大色系, 纤维颜色单一, 着色不均匀, 色牢度和色饱和度低[4], 这些问题在很大程度上限制了彩色棉的利用和发展, 因此, 亟待对彩色棉纤维色泽品质进行改良以提高其市场价值。
棕色棉纤维色素形成的前体物质是原花青素[5-6], 又称缩合丹宁, 是一类通过植物类黄酮次生代谢途径合成的聚多酚类化合物[5-7]。原花青素生物合成途径的研究已取得巨大进展, 已知经公共苯丙烷途径、核心类黄酮-花青素途径、原花青素特异途径生成原花青素的前体物质表儿茶素[8-10]。原花青素的前体物质是在细胞质中合成的, 需经过转运蛋白运输到液泡内聚合生成多聚体才能呈现出各种颜色[11]。MATE家族蛋白(multidrug and compound extrusion)在原花青素的转运过程中发挥着重要作用, 因此,基因可用作利用基因工程技术改良彩色棉纤维色泽品质的候选基因[12-13]。
MATE是一个新型的多药转运蛋白家族, 分布于原核生物、真核生物中, 大部分成员都有12个跨膜区域, 此类转运蛋白对氨基葡糖、阳离子染料、多种抗生素和药物具有转运作用[14-15]。研究者对拟南芥MATE家族的研究较为透彻, 拟南芥中至少有56个基因, 根据系统进化关系可分为7个亚家族, 分别在Fe3+和Al3+等金属离子、类黄酮、四甲胺、水杨酸等次级代谢物质转运方面发挥作用[16-17]。拟南芥基因编码一种类黄酮循环必需的MATE转运蛋白, 定位于液泡膜上, 它利用膜两侧的H+/Na+浓度梯度作为推动力, 将胞浆内合成的花青素、原花青素等多酚类色素的单体物质转运到液泡等亚细胞器官中[18-19]。蒺藜苜蓿能促进液泡吸收单宁的前体物质表儿茶素-3’-0-葡萄糖苷, 将转入拟南芥突变体, 可以使其种皮颜色恢复至野生型, 同时原花青素的含量也恢复至野生型水平, 表明是蒺藜苜宿中原花青素跨膜转运的关键基因[12,20]。苹果和在系统进化树中均与聚为一类, 将这2个基因分别转入拟南芥突变体, 均可以使突变体恢复至野生型, 表明和是苹果中转运原花青素的关键基因[21]。
迄今为止, 陆地棉中仅分离出2个MATE家族基因[6,22], 陆地棉基因组测序的完成使得MATE家族基因的鉴定工作成为可能。本研究鉴定陆地棉中的MATE家族基因, 全面、系统地解析基因的基本信息、保守域结构、进化关系、染色体定位以及组织表达分析等信息, 为进一步克隆和利用棉花基因改良棉纤维的色泽品质提供一定的理论基础。
1 材料与方法
1.1 陆地棉MATE家族基因的鉴定
以已克隆的陆地棉基因(CotAD_26128)的氨基酸序列作为查询(Query)序列, 利用BlastP从陆地棉全基因组数据库(https://www.cottongen.org/)中搜索MATE家族基因, 初步筛选出基因序列。用SMART (http://smart.emblheidelberg.de/)和Pfam (http://pfam.sanger.ac.uk/)软件鉴定陆地棉基因编码的蛋白序列是否存在跨膜结构, 设置参数为缺省值, 去除非全长的短片段以及相同基因的冗余序列, 保存完整且具有表达信息的基因序列用于下一步分析, 最终确定陆地棉基因组中MATE家族基因的序列。
1.2 MATE家族基因编码的氨基酸序列分析
利用ExPAsy (http://web.expasy.org/)在线工具分析陆地棉MATE家族蛋白的基本理化性质, 如氨基酸序列的等电点(pI)、蛋白质残基数(aa)。利用陆地棉基因组数据库(https://www.cottongen.org/)分析开放阅读性框(ORF)。利用Softberry (http://www. softberry.com/)和PSORT (http://psort.hgc.jp/form. html)对氨基酸序列进行亚细胞定位预测。
1.3 MATE家族基因进化树的构建
利用MEGA6.0提供的ClustalW程序对91个陆地棉和56个拟南芥的MATE蛋白进行多重序列比对, 然后通过邻位相连法(Neighbor-Joining, NJ, BootStrap = 1000)构建陆地棉和拟南芥中基因家族的复合进化树。从拟南芥网站(http://www. arabidopsis.org/)下载拟南芥MATE蛋白序列。
1.4 MATE家族基因的结构分析
采用DNAMAN和ClustalX对MATE家族蛋白氨基酸序列进行多重序列比对; 利用在线软件TMHMM (http://www.cbs.dtu.dk/services/)预测MATE蛋白的跨膜结构域(transmembrane domain, TM); 从陆地棉全基因组数据库下载MATE家族基因的外显子和内含子信息, 利用在线软件Gene Structure Display Server (GSDS) (http://gsds.cbi.pku.edu.cn/)分析陆地棉基因外显子–内含子(intron–exon)的数目及分布。
1.5 陆地棉MATE家族基因的染色体定位分析
从陆地棉全基因组数据库获得MATE家族基因的染色体定位信息, 以及陆地棉各条染色体的长度信息, 利用MapInspect软件绘制GhMATE家族基因的染色体定位图。
1.6 陆地棉MATE家族基因在棉花不同组织中的表达分析
1.6.1 植物材料的准备 陆地棉(L.)品种“新陆早33”、“新彩棉5号”由新疆石河子大学棉花所提供, 播种于石河子大学试验农场, 常规管理。在棉花盛花期挂牌标记当日开花的花蕾, 以第3天为时间节点, 摘取3~27 DPA (days post-anthesis, 开花后天数)棉铃, 室内剥取胚珠及纤维; 取棉花种子用浓硫酸脱绒后播种于花盆(营养土∶蛭石=2∶1)中, 28℃培养2~3周, 待幼苗长出2片真叶时将其拔出, 水培2 d之后取其根、茎、叶, 花从田间摘取。将上述植物材料液氮速冻后, 保存于–80℃冰箱, 用于提取RNA。
1.6.2 棉花总RNA的提取及cDNA第1链的合成
采用改良的CTAB酸酚法提取棉花样品总RNA[23], 用DNase I处理后利用Nanodrop1000紫外分光光度技测定OD260和OD280值, 计算RNA的浓度与纯度。用1.0%琼脂糖凝胶电泳检测RNA的完整性, 保存于–80℃备用。按照大连宝生物工程有限公司Reverse Transcriptase M-MLV (RNaseH-)说明书合成cDNA第1链。
1.6.3 陆地棉MATE家族基因的qRT-PCR分析
根据陆地棉MATE家族基因的cDNA序列, 采用Primer5.0软件在基因序列3′端特异性区域设计引物(表1), 扩增片段约为200 bp。分别以白色棉“新陆早33”、棕色棉“新彩棉5号”幼苗的根、茎、叶、花和不同发育时期棉纤维cDNA为模板, 利用qRT-PCR方法检测陆地棉MATE家族基因的表达情况, 内参基因为(GenBank登录号为DQ116441.1)。qRT-PCR反应体系为10.0 μL, 其中cDNA 1 μL、2×FASTSYBR混合物5.0 μL、基因特异正向引物(10 μmol L–1) 0.2 μL、反向引物(10 μmol L–1) 0.2 μL, RNase-Free H2O 3.6 μL。qRT-PCR在LightCycler 480II系统上进行, 反应程序为94℃预变性1 min; 95℃变性15 s, 56.5℃退火20 s, 72℃延伸30 s, 45个循环。按照2–∆∆Ct计算基因的相对表达值。实验中采集每种材料3组重复样品, 每个样品的RNA提取需进行3次重复, 共计9组实验结果, 计算其平均值和标准差。

表1 实验中所用的引物
2 结果与分析
2.1 陆地棉MATE家族基因的鉴定与命名
用已克隆的棕色棉MATE基因(CotAD_26128)为探针序列, 在陆地棉全基因组数据库(https:// www.cottongen.org/)中比对, 获得了91个候选MATE家族基因, 分别命名为(表2)。彩色棉MATE家族基因的编码序列(coding sequence, CDS)长1119~1632 bp, 编码374~548个氨基酸; 家族成员蛋白质的分子量介于36.6~60.3 kD之间, 理论等电点介于5.17~9.22之间。对各成员蛋白序列的亚细胞定位预测表明, 多数GhMATE (66个)被定位在细胞膜上, 少数被定位在液泡膜(8个)、叶绿体膜(7个)和细胞质膜(10个)上(表2)。
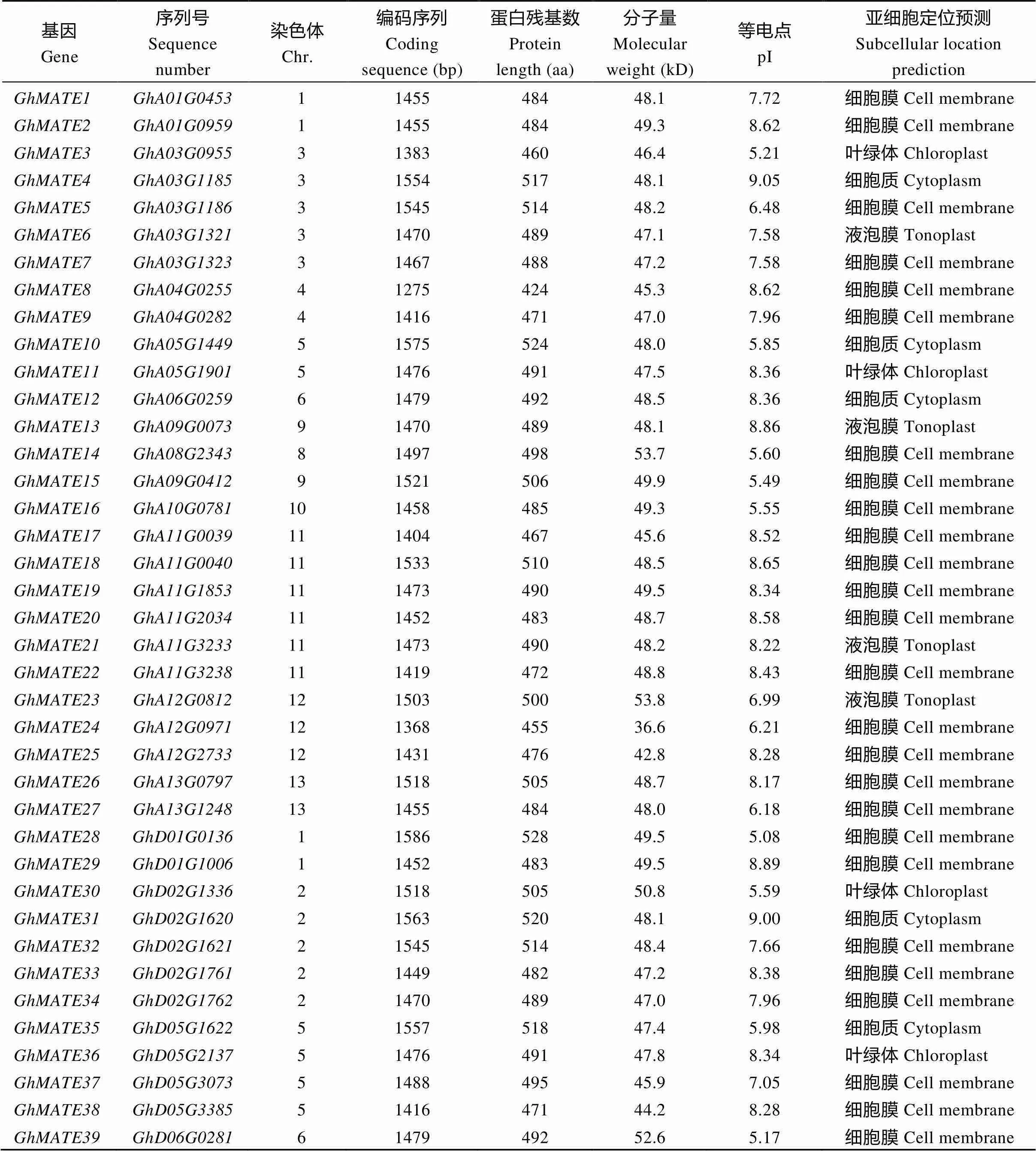
表2 陆地棉MATE蛋白家族成员的鉴定
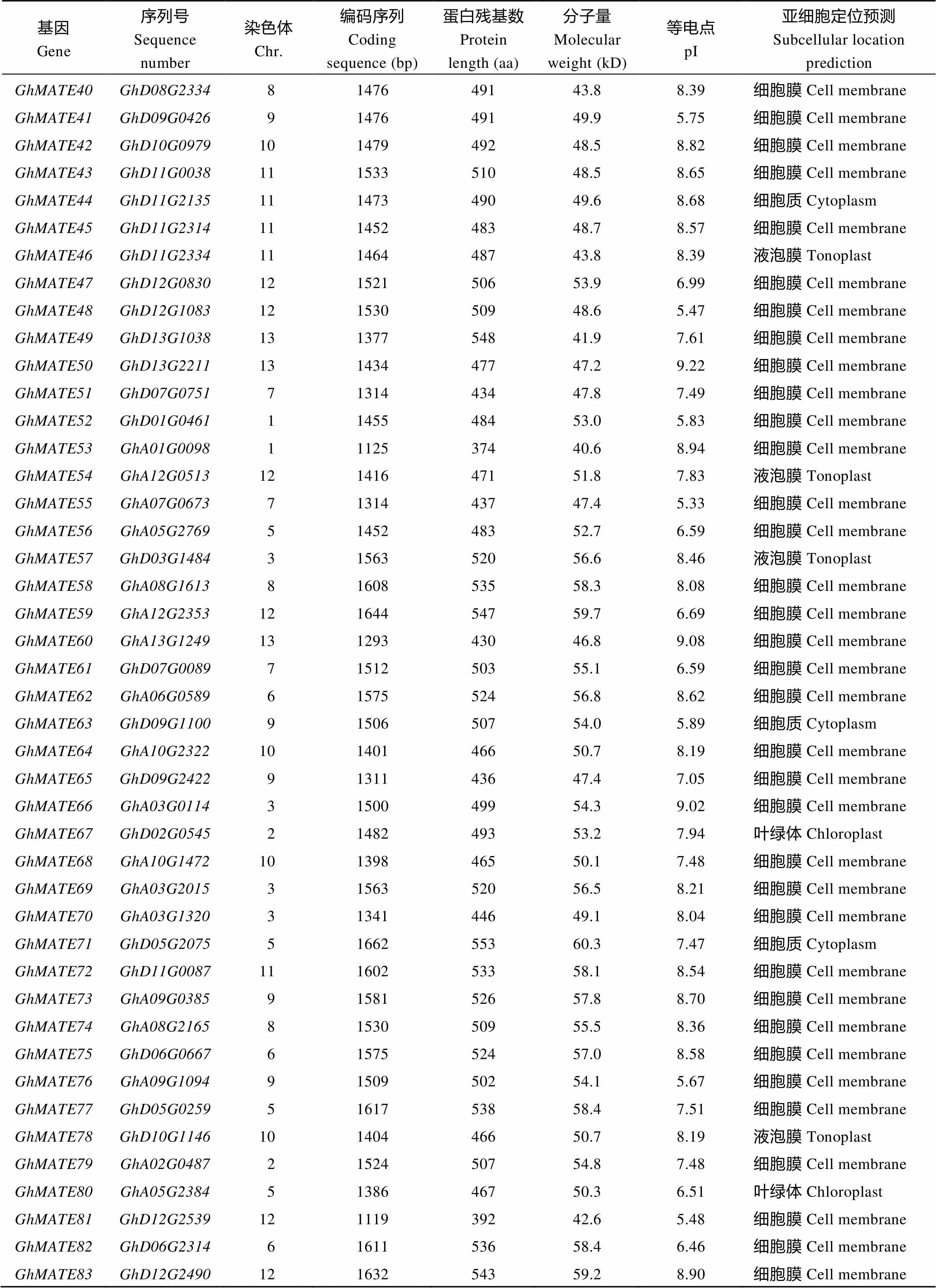
(续表2)

(续表2)
2.2 陆地棉MATE家族基因的进化关系分析
图1显示, 56个拟南芥MATE蛋白被清晰地分成A、B、C、D、E、F、G 7个亚家族, 陆地棉MATE蛋白被分至不同的亚家族中。其中A亚家族是最大的一个进化分支, 包括23个GhMATE家族成员, 其次是E、F和G亚家族各包括13个成员, B亚家族包括11个成员, C和D亚家族各包括9个成员。有13个基因和拟南芥基因聚在一个分支, 属于E亚家族。
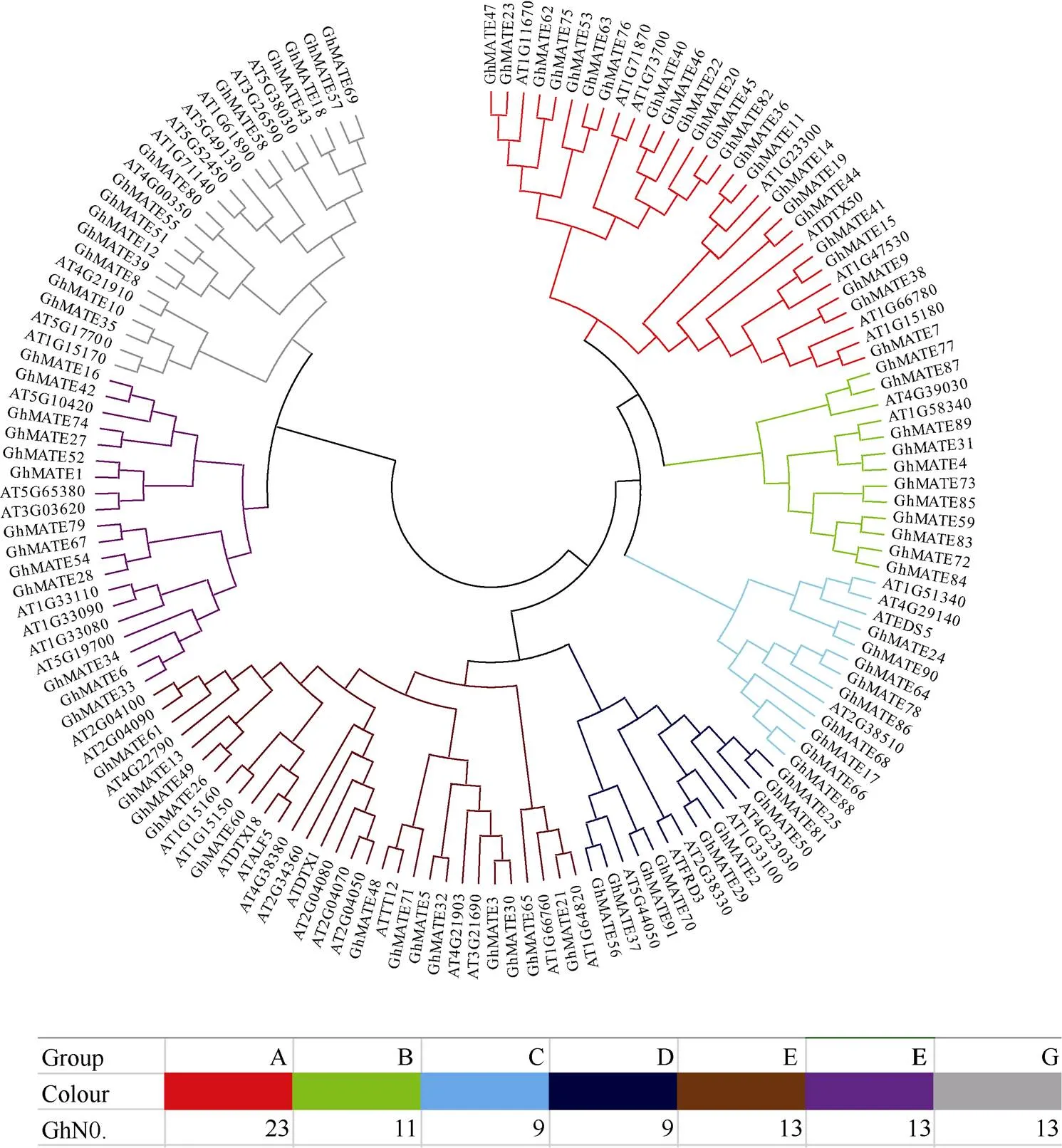
图1 陆地棉、拟南芥MATE家族蛋白成员进化分析
利用MEGA5.1软件及邻位相连法构建进化树, 设置Bootstrap参数为1000次重复, 有7个进化分支被命名为A~G, 7种不同的颜色代表7个亚家族, N0.代表每个分支陆地棉的成员。
The phylogenetic tree was generated using the neighbor-joining criteria in MEGA 5.1 with 1000 Bootstrap replicates; the seven distinct clades were named as A–G. The seven different color boxes represent seven clades. N0. representsmembers in each clade.
2.3 陆地棉MATE家族基因的保守域和结构分析
GhMATE家族成员含有10~13个TM, 其中GhMATE43蛋白含有13个TM, GhMATE25、GhMATE67、GhMATE70和GhMATE73含有11个TM, GhMATE24、GhMATE90含有10个TM, 剩余84个GhMATE蛋白均含有12个TM, 部分GhMATE蛋白的12个跨膜结构域如图2所示。对获得的91个GhMATE家族基因的内含子和外显子的个数和排布情况分析表明, 大多数基因(50个)由7~8个外显子和6~7个内含子组成, 其中27个基因含有8个外显子和7内含子, 23个基因含有7个外显子和6个内含子; 23个基因仅由外显子组成, 不含有内含子, 这些基因分别存在于B、C、D、E、F和G亚家族中, 其中C和D亚家族中居多; 剩余的18个基因中, 3个基因由9个外显子和8个内含子组成, 其他15个基因由2~6个外显子和1~5个内含子组成(图3)。

图2 不同亚家族中GhMATE蛋白的多重序列比对

图3 陆地棉MATE基因的进化树和基因结构比较
蓝色表示UTR (上下游序列), 绿色表示编码区, 灰线表示内含子区。
The blue shows the UTR, the green indicates the exon, and the gray indicates the intron.
2.4 陆地棉MATE家族基因的染色体定位
91个GhMATE家族成员分布在25条染色体上, 其中48个分布在13个A基因组上, 43个分布在12个D基因组上, Chr.D4染色体上未发现基因(图4)。染色体Chr.A3上分布的基因最多, 为8个; 其次是Chr.A11, 为7个, Chr.D2、Chr.D5和Chr.D11上各分布6个; 染色体Chr.A5、Chr.A9和Chr.A12上各分布5个; 染色体Chr.A2、Chr.D9和Chr.D12上各分布4个; 其余14个染色体上各分布1~3个。根据200 kb的核苷酸中含有3个以上基因即为1个基因簇的定义[24], GhMATE家族基因在3条染色体上形成了1个基因簇, 分别为Chr.A11 (和)、Chr.D2 (和)和Chr.D11 (和)。在Chr.A3染色体上形成了2个基因簇, 即形成1个,形成了另1个, 其他染色体上未形成基因簇。
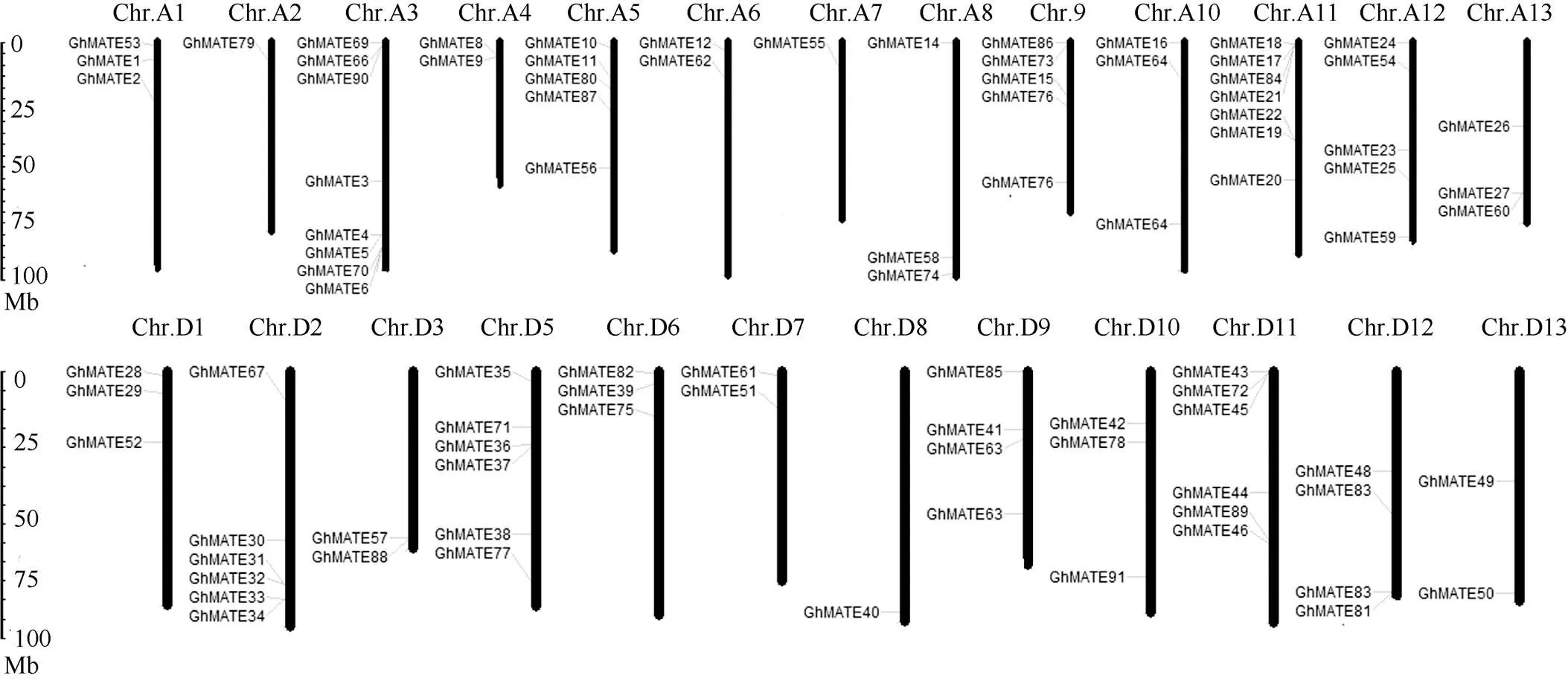
图4 陆地棉MATE家族基因染色体定位
2.5 陆地棉MATE家族基因的表达模式分析
从7个亚家族中分别选取20多个基因进行表达模式分析, 包括所有定位于液泡膜上的基因、13个E亚家族成员以及随机选取的其他亚家族成员。qRT-PCR结果发现, GhMATE家族基因在白色棉、棕色棉不同器官(根、茎、叶、花)及棉纤维细胞的不同发育时期均有表达。根据qRT-PCR结果, 从中筛选出、、、、、和共7个在棕色棉纤维中优势表达的基因, qRT-PCR结果显示,和基因在棕色棉纤维中的表达量明显高于白色棉, 在棕色棉纤维中的表达量变化呈现先升高后降低的趋势, 分别在15DPA和21DPA达到最大值, 几乎是白色棉纤维表达量的7~10倍。、和基因在棕色棉纤维中的表达量均高于白色棉纤维, 均在21 DPA时达到最大值, 但仅为白色棉表达量的1.0~1.8倍。和在15~27 DPA的棕色棉纤维中的表达量高于白色棉纤维, 但两者表达量的倍数较低。在棉纤维发育过程中, 上述7个基因在棕色棉纤维中表达量均高于白色棉, 其中和在棕色棉纤维中的表达优势最明显, 暗示它们可能与棕色棉纤维的颜色形成相关。
3 讨论
MATE蛋白是一个新型的多药转运蛋白家族, 其成员数量庞大, 大概有203个已经测序的基因广泛分布于古细菌、细菌、酵母、动物和植物中[13-15], 如拟南芥中58个、葡萄中57个、杨树中58个、玉米和短柄草中38个[25]。模式植物拟南芥和烟草中MATE家族基因的复杂相关特征和生物学功能已被挖掘和鉴定[17,26]。拟南芥中MATE转运蛋白可分为A、B、C、D、E、F和G亚家族, 分别在Fe3+和Al3+等金属离子、类黄酮、四甲胺、水杨酸等次级代谢物质转运方面发挥作用。A亚家族的(At3g23560)与转运四甲铵毒素物质相关[27]; C亚家族的(At4g39030)可能与转运水杨酸或其前体物质相关[28]; D亚家族的(At3g08040)具有维持拟南芥内环境铁离子动态平衡功能[29]; E亚家族的(At2g04070)在植物体内介导生物碱、抗生素和其他有毒化合物转出[30];(At3g59030)定位在液泡膜上, 与原花青素的前体物质向液泡的转运有关[16-17]。研究者从彩色棉中分离到4个黄酮类基因, 共获得5个色素合成相关基因和, 并据此推测棕色棉中的色素物质是原花青素[31]。原花青素是在细胞质中合成的, 再经转运蛋白运输到液泡中。MATE家族蛋白在花青素和原花青素的转运过程中起重要作用[12-13]。本研究在彩色棉属棕色棉中鉴定出91个GhMATE家族成员, 分为7个亚家族, 其中13个棕色棉基因与拟南芥原花青素转运相关基因聚为一类, 表明基因可能参与细胞内原花青素向液泡的转运, 从而影响彩色棉纤维颜色的形成。

图5 MATE家族基因在棉花不同组织中的qRT-PCR分析
R: 根; S: 茎; L: 叶; F: 花; 6~27 DPA:开花后6、9、12、15、18、21、24、27 d纤维。
R: root; S: stem; L: leaf; F: flower; 6–27 DPA: fiber cells at 6, 9, 12, 15, 18, 21, 24, and 27 DPA, respectively.
转运蛋白的亚细胞定位对其行使生物学功能至关重要, 不同的亚细胞定位可能引起转运蛋白功能的差异。植物MATE蛋白主要定位于质膜或液泡膜[32]。转运原花青素的MATE转运蛋白大多定位在液泡膜上, 将原花青素的前体物质糖基化/酰基化的表儿茶素转运至液泡中[11,33]。本研究表明, 多数定位在细胞膜上, 少数定位在液泡膜, 叶绿体膜和细胞质膜上。8个定位在液泡膜上的基因可能与原花青素的前体物质糖基化/酰基化表儿茶素的转运有关, 使原花青素在液泡中积累、聚合形成不同聚合度的原花青素聚合物, 从而决定彩色棉纤维的颜色。由于E亚家族的基因和定位于液泡膜的基因可能与原花青素的转运相关, 推测它们可能存在较高的一致性, 比较后发现, 两组基因仅有一个重合的基因, 说明研究者对植物MATE家族基因的结构和功能研究尚不完善, 其他亚家族成员中也可能存在与原花青素转运相关的基因。
MATE蛋白家族长度在400~700个氨基酸之间, 大多数氨基酸成员包括400~550个残基[34]。本研究中GhMATE家族成员编码374~548个氨基酸残基, 符合MATE家族成员的基本特征。根据前人研究, MATE转运蛋白约有12个TM, 如拟南芥MATE蛋白家族含有8~13个TM, FRD3含有13个, EDS5含有11个TM[35]。12个TM是植物MATE蛋白行使功能的核心元件[36]。本研究发现84个蛋白序列均含有12个TM, 1个蛋白含有13个TM, 5个蛋白少于12个TM。12个TM符合MATE转运蛋白的基本特征, 表明大多数GhMATE蛋白均具有行使蛋白功能的完整核心元件。
基因在棕色棉、白色棉不同组织和棉纤维细胞的不同发育时期均有表达, 从7个亚家族中共找到7个在棕色棉纤维中优势表达的基因, 其中和在棕色棉纤维中的表达量明显高于白色棉, 且都定位于液泡膜上。拟南芥TT12是第一个被发现定位于液泡膜上, 参与原花青素前体表儿茶素3′-葡萄糖苷的转运蛋白[12,18,33]。苜蓿定位在液泡膜上, 优先运输原花青素合成前体表儿茶素3’-O-葡糖苷, 能够弥补拟南芥突变体种子的表型缺陷[12,20]。葡萄中也鉴定出2个与MATE家族序列同源性很高的基因和, 在花青素生物合成的组织中表达, 特异运输酰基化的花青素[37]。Frank等[21]在苹果中克隆了2个基因, 命名为和, 系统进化分析表明, 它们与聚为一类, 这2个基因均能使拟南芥突变体恢复至野生型, 证明和是苹果中转运原花青素的关键基因[16]。本研究通过系统发育分析, 发现GhMATE13蛋白与拟南芥原花青素转运相关蛋白聚为一类, 且都定位在液泡膜上, 表明GhMATE13可能参与原花色素的转运及液泡积累, 在彩色棉纤维颜色形成过程中起重要作用。只定位于液泡膜上, 属于A亚家族成员, 推测在棕色棉颜色形成过程中可能发挥着重要作用, 其功能有待进一步研究。本研究有助于进一步研究GhMATE家族基因在棕色棉纤维颜色形成中的作用机制。
4 结论
鉴定出91个GhMATE家族基因, 将它们分为A、B、C、D、E、F和G 7个亚家族, 其中84个GhMATE蛋白具有12个典型的跨膜结构域, GhMATE家族成员被定位在25条染色体上, 共形成5个基因簇。和在棕色棉纤维中的表达量明显高于在白色棉中, 可能在棕色棉纤维的颜色形成过程中发挥重要作用。
[1] 赵向前, 王学德. 天然彩色棉纤维色素成分的研究. 作物学报, 2005, 31: 456–462Zhao X Q, Wang X D. Study on pigment constituents of natural colored cotton fiber., 2005, 31: 456–462 (in Chinese with English abstract)
[2] 邱新棉, 周文龙, 李茂松, 马永根. 天然彩色棉纤维色素的遗传基础形成及湿处理色素变化规律的研究. 中国农业科学, 2002, 35: 610–615Qiu X M, Zhou W L, Li M S, Ma Y G. Study on the changes of pigment and wet processing to form the genetic basis of natural color cotton fiber pigment., 2002, 35: 610–615 (in Chinese with English abstract)
[3] 董合忠, 李维江, 唐薇, 张冬梅. 彩色棉纤维发育与色素形成. 中国棉花, 2004, 31(2): 2–4Dong H Z, Li W J, Tang W, Zhang D M. Pigment synthesis and cotton fiber development of color cotton.2004, 31(2): 2–4 (in Chinese with English abstract)
[4] 赵兴华, 渠云芳, 黄晋玲. 彩色棉育种研究现状与展望. 现代农业科技, 2011, (5): 84–85Zhao X H, Qu Y F, Huang J L. Research status and prospect of colored cotton breeding., 2011, (5): 84–85 (in Chinese with English abstract)
[5] Li Y J, Zhang X Y, Wang F W, Yang C L, Liu F, Xia G X, Sun J. A comparative proteomic analysis provides insights into pigment biosynthesis in brown colored fiber., 2013, 78: 374–388
[6] Feng H J, Li Y J, Wang S F, Liu Y C, Xue F, Zhang L L, Sun J. Molecular analysis of proanthocyanidins related to pigmentation in brown cotton fibre (L.)., 2014, 65: 5759–5769
[7] Akagi T, Ikegami A, Suzuki Y, Yoshida J, Yamada M, Sato A, Yonemori K. Expression balances of structural genes in shikimate and flavonoid biosynthesis cause a difference in proanthocyanidin accumulation in persimmon (Thunb.) fruit., 2009, 230: 899–915
[8] Pourcel L, Irani N G, Lu Y, Riedl K, Schwartz S and Grotewold E. The formation of anthocyanic vacuolar inclusions inand implications for the sequestration of anthocyanin pigments., 2010, 3: 78–90
[9] Xu W, Grain D, Bobet S, Gourrierec J, Thevenin J, Kelemen Z, Lepiniec L, Dubos C. Complexity and robustness of the flavonoid transcriptional regulatory network revealed by comprehensive analyses of MYB-bHLH-WDR complexes and their targets in Arabidopsis seed., 2014, 202: 132–144
[10] Kitamura S, Matsuda F, Tohge T, Yonekura-Sakakibara K, Yamazaki M, Saito K, Narumi I. Metabolic profiling and cytological analysis of proanthocyanidins in immature seeds offlavonoid accumulation mutants., 2010, 62: 549–559
[11] Pang Y Z, Abeysinghe I S B, He J, He X Z, Huhman D, Mewan K M, Sumner L W, Yun J F, Dixon R A. Functional characterization of proanthocyanidin pathway enzymes from tea and their application for metabolic engineering., 2013, 161: 1103–1116
[12] Zhao J, Dixon R A. MATE transporters facilitate vacuolar uptake of epicatechin 3’-0-glucoside for proanthocyanidin biosynthesis inand Arabidopsis., 2009, 21: 2323–2340
[13] Kleindt C K, Stracke R, Mehrtens F, Weisshaar B. Expression analysis of flavonoid biosynthesis genes during Arabidopsis thaliana silique and seed development with a primary focus on the proanthocyanidin biosynthetic pathway., 2010, 3: 255
[14] He X, Szewczyk P, Karyakin A, Evin M, Hong W X, Zhang Q. Structure of a cation-bound multidrug and toxic compound extrusion transporter., 2010, 467: 991–994
[15] Tiwari M, Sharma D, Singh M, Tripathi R D, Trivedi P K. Expression ofandalters development, stress responses and pathogen susceptibility in Arabidopsis., 2014,4: 3964
[16] Roschzttardtz H, Seguela-Arnaud M, Briat J F, Vert G, Curie C. The FRD3 citrate effluxer promotes iron nutrition between symplastically disconnected tissues throughout Arabidopsis development., 2011, 23: 2725–2737
[17] Thompson E P, Wilkins C, Demidchik V, Davies J M, Glover B J. An Arabidopsis flavonoid transporter is required for anther dehiscence and pollen development., 2010, 61: 439–451
[18] Debeaujon I, Peeters A J M, Leon-Kloosterziel K M, Koornneef M. Thegene of Arabidopsis encodes a multidrug secondary transporter-like protein required for flavonoid sequestration in vacuoles of the seed coat endothelium., 2001, 13: 853–871
[19] Pourcel L, Routaboul J M, Kerhoas L, Caboche M, Lepiniec L, Debeaujon I.encodes alaccase-like enzyme involved in oxidative polymerization of flavonoids in Arabidopsis seed coat.2005, 17: 2966–2980
[20] Zhao J, Huhman D, Shadle G, He X Z, Sumner L W, Tang Y H, Dixon R A. MATE2 mediates vacuolar sequestration of flavonoid glycosides and glycoside malonates in., 2011, 23: 1536–1555
[21] Frank S, Keck M, Sagasser M, Niehaus K, Weisshaar B, Stracke R. Two differentially expressed MATE factor genes from apple complement the Arabidopsis transparentmutant., 2011, 13: 42–50
[22] 冯鸿杰. 天然彩色棉纤维色素物质的鉴定及其代谢机制研究. 石河子大学博士学位论文, 新疆石河子, 2014 Feng H J. Identification and Metabolic Mechanism of Pigment Fiber in Natural Colored Cotton. PhD Dissertation of Shihezi University, Shihezi, Xinjiang, China, 2014 (in Chinese with English abstract)
[23] 蒋建雄, 张天真. 利用CTAB/酸酚法提取棉花组织总RNA. 棉花学报, 2003, 15: 166–167 Jiang J X, Zhang T Z. Extraction of total RNA in cotton tissues with CTAB-acidic phenolic method., 2003, 15: 166–167 (in Chinese with English abstract)
[24] Winzer T, Gazda V, He Z, Kaminski F, Kern M, Larson T R, Li Y, Meade F, Teodor R, Vaistij F E, Walker C, Bowser T A, Graham I A. A10-gene cluster for synthesis of the anticancer alkaloid noscapine., 2012, 336: 1704–1708
[25] Brown M H, Paulsen I T, Skurray R A. The multidrug efflux protein NorM is a prototype of a new family of transporters., 1999, 31: 394–395
[26] Shitan N, Minami S, Morita M, Hayashida M, Ito S, Takanashi K, Omote H, Moriyama Y, Sugiyama A, Goossens A. Involvement of the leaf-specific multidrug and toxic compound extrusion (MATE) transporter Nt-JAT2 in vacuolar sequestration of nicotine in, 2014, 9(9): e108789
[27] Diener A C, Gaxiola R A, Fink G R. Arabidopsis ALF5, a multidrug efflux transporter gene family member, confers resistance to toxins., 2011, 13: 1625–1638
[28] Yamasaki K, Motomura Y, Yagi Y, Nomura H, Kikuchi S, Nakai M, Shiina T. Chloroplast envelope localization of EDS5, an essential factor for salicylic acid biosynthesis in.2013,8(4): e23603
[29] Durrett T P, Gassmann W, Rogers E E. The FRD3-mediated efflux of citrate into the root vasculature is necessary for efficient iron translocation., 2007, 144: 197–205
[30] Zhang H, Zhu H, Pan Y, Yu Y, Luan S, Li L. ADTX/MATE type transporter facilitates abscisic acid efflux and modulates ABA sensitivity and drought tolerance in.,2014, 7: 1522–1532
[31] Xiao Y H, Zhang Z S, Yin M H, Luo M, Li X B, Hou L, Pei Y. Cotton flavonoid structural genes related to the pigmentation in brown fibers.2007, 358: 73–78
[32] Seo P J, Park J, Park M J, Kim Y S, Kim S G, Jung J H. A golgi- localized MATE transporter mediates iron homoeostasis under osmotic stress in., 2012, 442: 551–561
[33] Marinova K, Pourcel L, Weder B, Schwarz M, Barron D, Routaboul J M, Debeaujon I, Klein M. The Arabidopsis MATE transporter TT12 acts as a vacuolar flavonoid/H+-antiporter active in proanthocyanidin-accumulating cells of the seed coat., 2007, 19: 2023–2038
[34] Yazaki K, Sugiyama A, Morita M, Shitan N. Secondary transport as an efficient membrane transport mechanism for plant secondary metabolites., 2008, 7: 513–524
[35] Li L, He Z, Pandey G K, Tsuchiya T, Luan S. Functional cloning and characterization of a plant efflux carrier for multidrug and heavy metal detoxification., 2002, 277: 5360–5368
[36] Hvorup R N, Winnen B, Chang A B, Jiang Y, Zhou X F, Saier M H. The multidrug, oligosaccharidyl-lipid/polysaccharide (MOP) exporter superfamily., 2007, 270: 799–813
[37] Gomez C, Terrier N, Torregrosa L, Vialet S, Fournier-Level A, Verries C, Souquet J M, Mazauric J P, Klein M, Cheynier V. Grapevine MATE-type proteins act as vacuolar H+-dependent acylated anthocyanin transporters., 2009, 150: 402–415
Identification and Expression Analysis of Multidrug and Toxic Compound Extrusion Protein Family Genes in Colored Cotton
WANG Zuo-Min1, LIU Jin1, SUN Shi-Chao2, ZHANG Xin-Yu2, XUE Fei2, LI Yan-Jun2,*, and SUN Jie2
1College of Life Sciences, Shihezi University, Shihezi 832003, Xinjiang, China;2Agricultural College / Key Laboratory of Oasis Eco-agriculture Xinjiang Production and Construction Group, Shihezi University, Shihezi 832003, Xinjiang, China
MATE (multidrug and toxic compound extrusion) is a transport proteins family that can transport toxins, glucosamine, antibiotics and drugs. In this study, the MATE family genes were identified from genome database ofL. through bioinformatics analysisand phylogenetic relationship, chromosome distribution, gene structure and expression patterns of this family were comprehensively compared. A total of 91genes were found in the cotton genome and named as–.genes of cotton were classified into seven groups namely A, B, C, D, E, F, and G with a classification is consistent with Arabidopsis. Multiple sequence alignment and conserved domain prediction indicated that 84 of the 91 GhMATE proteins contained 12 typical transmembrane domains. The chromosome mapping analysis showed thatgenes were distributed with different densities over 25 chromosomes and clustered into five clusters. The qPCR showed that thegenes expressed in all tissues of cotton with different expression patterns.andwere preferentially expressed in brown cotton fibers than in white cotton fibre, suggesting that they may play an important role in brown color formation of cotton fibre. This study provides valuable informations for dissecting functions and molecular mechanisms ofin cotton.
L.; naturally colored cotton; MATE family; bioinformatics; qRT-PCR (quantitative real-time PCR)
2017-11-01;
2018-04-11;
2018-05-10.
10.3724/SP.J.1006.2018.01380
李艳军, E-mail: lyj20022002@sina.com.cn
E-mail: 1427786863@qq.com
本研究由国家自然科学基金项目(U1303281)和石河子大学动植物育种重点项目(YZZX201601, gxjs2014-yz08)资助。
This study was supported by the National Natural Science Foundation of China (U1303281) and the Key Projects of Plant and Animal Breeding of Shihezi University (YZZX201601, gxjs2014-yz08).
URL:http://kns.cnki.net/kcms/detail/11.1809.S.20180508.1644.006.html
