lprG基因表达上调对THP1细胞表达谱的影响
2018-09-10宛宝山陈伟伟张冉冉
曹 雯, 宛宝山, 陈伟伟, 张冉冉, 乐 军, 陈 晋
(同济大学附属上海市肺科医院检验科,上海 200433)
结核分枝杆菌(Mycobacteriumtuberculosis,Mtb)是结核病(tuberculosis,TB)的病原菌,感染机体后主要寄居在巨噬细胞内。巨噬细胞作为抵抗Mtb的第一道防线,是先天性免疫和适应性免疫联系的桥梁[1]。Mtb感染机体后,巨噬细胞主要通过细胞膜表面的Toll样受体(Toll-like receptors, TLRs)识别Mtb细胞表面的膜成分,如脂蛋白、糖脂、甘露糖和海藻糖等,经内吞作用介导Mtb进入巨噬细胞内,加工处理Mtb后提呈给CD4+T细胞,进而触发机体的免疫应答反应[2-3]。脂蛋白(lipoprotein, Lpp)是由Mtb合成并经过脂肪修饰后分泌到胞外锚定在细胞膜上的一种蛋白,其特征是有一个N-端分泌信号肽。脂蛋白在Mtb中表达十分丰富,生物信息学预测Mtb有99个编码脂蛋白的基因,约占蛋白组中预测蛋白的2.5%[4-5]。lprG是相对分子质量为27000的脂蛋白,与Mtb的毒力相关,是制备减毒活疫苗的候选因子[6-8]。lprG作为TLR2受体的激动剂,能够调节机体免疫应答使得Mtb躲避免疫攻击[9]。近年来,关于lprG与巨噬细胞相互作用的研究得到广泛关注,本实验通过建立稳定表达lprG的THP1细胞系(人单核源性巨噬细胞系),模拟lprG与宿主巨噬细胞相互作用模型,利用RNA-seq技术筛选差异表达基因,采用GO及KEGG通路分析进行生物信息学分析差异基因,为进一步研究在Mtb感染宿主过程中,lprG与巨噬细胞的相互作用提供基础。
1 材料与方法
1.1 质粒与细胞
慢病毒表达质粒pGMLV-CMV-EF1-ZsGreen1-T2A-Puro和包装质粒购自上海吉满生物科技有限公司;HEK293T和THP1细胞购自上海生命科学院。
1.2 主要试剂
PrimeSTAR®HS DNA Polymerase、DH5α购自日本TaKaRa公司;XhoI和EcoRI内切酶、T4 DNA Ligase、DNA Ladder购自加拿大Fermentas公司; 质粒小抽试剂盒、琼脂糖凝胶DNA回收试剂盒购自上海捷瑞生物工程有限公司;LipofectaminTM2000、TRIzol、Superscript VILO cDNA Synthesis Kit购自美国Invitrogen公司;FBS、DMEM、青链霉素、胰酶购自美国Thermo Fisher公司;慢病毒浓缩试剂盒购自上海吉满生物科技有限公司;无内毒素质粒大抽试剂盒购自德国Qiagen公司;SYBR Green PCR Master Mix购自美国Applied Biosystems公司;Illumina®TruSeq®RNA Sample Preparation Kit v2、TruSeq PE Cluster Kit购自美国Illumina公司。
1.3 方法
1.3.1 引物设计 根据GenBank设计目的基因引物。lprG上游引物序列为5′-CCGGAATTCGCCACCA-TGCATCATCACCATCACCATCG-3′,下游引物序列为5′-CCGCTCGAGCTTATCGTCGTCATCCTTG-TAATCGC-3′,由生工生物工程(上海)股份有限公司合成。
1.3.2 慢病毒表达质粒的构建 慢病毒载体pGMLV-CMV-EF1-ZsGreen1-T2A-Puro和目的片段经过EcoRI和XhoI双酶切回收后在16℃下过夜连接,鉴定筛选阳性菌落后送往生工生物工程(上海)股份有限公司合成进行测序。
1.3.3 慢病毒的包装及滴度测定 将空载质粒和测序正确的重组质粒各取10μg与20μg包装质粒共同转染293T细胞进行慢病毒包装,培养至48、72h收集病毒上清液,按照浓缩试剂盒说明书进行浓缩后保存。
1.3.4 梯度稀释法测定慢病毒滴度 将细胞均匀铺在96孔板中,每个病毒准备10个离心管,加入90μL完全培养基,第1个管中加入10μL病毒液,混匀后再吸取10μL至第2个管中混匀,依次类推,做10个稀释度后感染细胞。培养至第5天在荧光显微镜下观察各孔中荧光细胞数量,计算慢病毒滴度,滴度符合实验条件后进行后续的细胞感染。
1.3.5 慢病毒感染及筛选THP1稳转细胞系 第1天后,将THP1悬浮细胞培养在24孔板中,培养基中含慢病毒和5μg/mL嘌呤霉素,每孔体积为200~300μL,不离心感染。72h后观察感染效率,待感染成功后继续用含5μg/mL嘌呤霉素的新鲜培养基筛选至获得稳转细胞系: 稳定表达lprG的THP1细胞系(lprG-THP1)和空载的THP1细胞系(NC-THP1)。
1.3.6 qPCR及Western印迹法检测lprG在THP1细胞中的表达 用TRIzol法提取两组样品中的总RNA,用Agilent 2100生物分析仪对RNA进行质检。按照RNA 2μL、5×VILO Reaction Mix 4μL、10×SuperScript Enzyme Mix 2μL、DEPC水12μL体系进行反转录cDNA,qPCR检测lprG的表达量。lprG引物序列如下。上游引物为5′-GACGGTCAACG-GCAAGTCC-3′ 、下游引物为5′-GAACACCACGA-AGTCGGCATC-3′;内参基因GAPDH引物序列: 上游引物为5′-GTCTCCTCTGACTTCAACAGCG-3′、下游引物为5′-ACCACCCTGTTGCTGTAGCCAA-3′。根据qPCR反应曲线得到lprG和GAPDH的Ct值,采用ΔΔCt的方法进行相对定量,根据F=2-ΔΔCt计算lprG的表达差异倍数。
Western印迹法: 将两组样品细胞放置在冰上,每孔加1 mL PBS清洗2次后,加100 μL RIPA裂解液冰上裂解后将裂解物转移至离心管内离心(离心半径为7.7cm,12000r/min,离心2min),取上清液。在上清液中加入5×的SDS-PAGE上样缓冲液20μL,99℃水浴加热10min,4℃离心10min后进行Western印迹法检测,实验条件为: 80V和120V电压下电泳30min和80min,400mA电流下转膜60min,TBST洗涤10min,含5%脱脂牛奶的TBST室温封闭2h,TBST洗涤3次,10min/次,4℃一抗(1∶1500稀释)孵育过夜,TBST洗涤3次,10min/次,二抗(1∶4000稀释)室温孵育2h,ECL化学发光法显影。
1.3.7 转录组测序与qPCR验证 对两组样品中的总RNA用Illumina®TruSeq®RNA Sample Preparation Kit v2构建cDNA文库。取10ng的cDNA文库,用TruSeq PE Cluster Kit在cBot系统中进行簇生成反应,然后在Illumina HiseqTM2500中进行双向测序,每组样品做3次技术重复,取3组数值的平均值以获得原始数据。使用FASTX-Toolkit软件(http:∥hannonlab.cshl.edu/fastx_toolkit/)对原始数据进行处理,去除低值序列得到过滤后的序列数,然后比对到拼接的基因数据库上,统计每个基因数据库中分别来自两个样品的序列数目,转换为PRKM,利用DEGseq程序包中的MARS模型,计算每个基因在两个样品中的表达丰度差异,FDR<0.001为差异有统计学意义。并对差异表达基因做了GO和KEGG通路显著性富集分析。选取6个基因C7、IL17C、MSR1、IL36A、WIF1和CTNNA2进行qPCR验证,采用ΔΔCt的方法进行相对定量,根据F=2-ΔΔCt计算表达差异倍数,引物序列见表1。
1.4 统计学处理
2 结 果
2.1 重组载体构建及鉴定
对重组慢病毒载体进行PCR产物鉴定,片段为801bp,与目的基因片段大小一致,对比测序结果与目的基因序列,结果一致。
2.2 慢病毒滴度
经过梯度稀释法检测,在荧光显微镜下观察6号孔中约有20个表达GFP的293T细胞,含有10×10-8mL慢病毒液,根据公式计算慢病毒滴度=20TU/(10×10-8)mL=2×108TU/mL,病毒滴度符合实验要求。
2.3 总RNA检测结果
提取NC-THP1和lprG-THP1样品中总RNA,在Agilent 2100生物分析仪中检测,浓度分别为71.4ng/μL和92.4ng/μL,RNA完整性数目(RNA integrity number, RIN)分别为9.8和10,RNA质量检测合格。
2.4 荧光观察及qPCR和Western印迹法检测目的基因lprG在THP1细胞中的表达
荧光显微镜观察到GFP在THP1细胞中表达,表明慢病毒成功侵染THP1细胞;qPCR检测到lprG在THP1细胞中过表达,Western印迹法检测到THP1胞质中有抗Flag信号,提示有lprG的表达。上述实验表明成功建立稳定表达lprG的THP1细胞系,见图1~3。

图1 荧光显微镜观察GFP在THP1细胞中表达(×100)Fig.1 The expression of GFP in THP1 cells observed with fluorescence microscope(×100)A: 明场 B: 荧光场
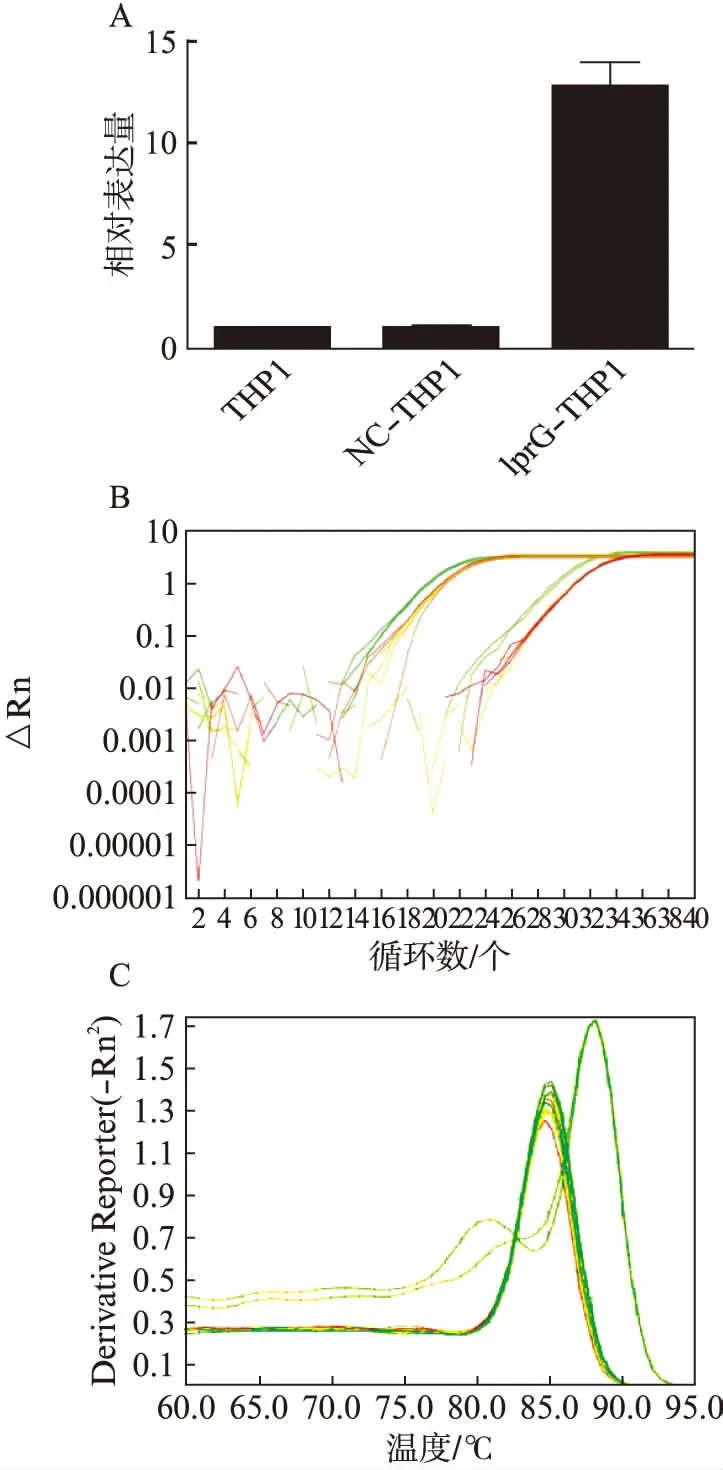
图2 qPCR检测THP1细胞中lprG的表达Fig.2 The expression of lprG in THP1 cells identified by qPCRA: 相对表达量 B: 扩增曲线 C: 熔解曲线
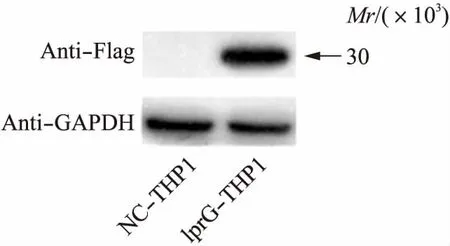
图3 Western印迹法检测THP1细胞中lprG的表达Fig.3 The expression of lprG in THP1 cells identified by Western blotting
2.5 转录组结果分析
对两组样品测序原始数据去除低值序列后,NC-THP1组和lprG-THP1组的过滤后的序列数分别为44和40,Q30 base分别为96.32%和95.57%。与NC-THP1组相比,lprG-THP1组共有712个基因表达有显著性差异,其中419个基因上调,占58.8%,293个基因下调,占41.2%。对两组样品差异基因进行GO分析,分为生物过程、分子功能和细胞组分3类。在生物过程中,主要为氨基酸及其衍生物代谢,占75.5%,其次为细胞内生物过程,占23.8%。在分子功能中,主要为蛋白质结合,占40.3%,其次为催化活性,占12.04%。在细胞成分中,主要为细胞内成分,占49.2%。通过对KEGG通路的分析发现有41个通路在差异表达基因中富集,其中有56个基因参与免疫应答及感染性疾病过程,见图4。

图4 免疫应答相关的差异表达基因Fig.4 The different expression of genes associated with immune response
2.6 qPCR验证结果
通过qPCR方法对6个差异表达基因(IL36A、MSR1、WIF1、IL17C、C7和CTNNA2)进行验证,得到lprG-THP1组与NC-THP1组基因表达量的比值,结果显示IL36A、MSR1、WIF1、IL17C和C7表达均上调,CTNNA2表达下调,与RNA-seq的结果相比较,6个基因的差异表达趋势一致,证明了RNA-seq的结果可靠,见图5。
3 讨 论
研究[10-11]表明,lprG能够通过TLR2途径抑制巨噬细胞MHC-Ⅱ抗原处理过程及TNF-α的产生,从而影响CD4+T细胞对Mtb的识别,导致机体免疫应答受损。另有研究[12-13]表明,lprG能阻碍巨噬细胞内吞噬体的成熟及吞噬体和溶酶体的结合,Mtb因此避开了蛋白酶的降解,得以长期寄存在巨噬细胞内生长繁殖。此外,lprG还参与体内三酰甘油的转运过程,通过调节巨噬细胞内三酰甘油的水平影响结核分枝杆菌的生长速度[14]。由此证明,lprG在免疫调节方面发挥着重要作用,其与巨噬细胞的相互作用影响着结核病的发生发展。
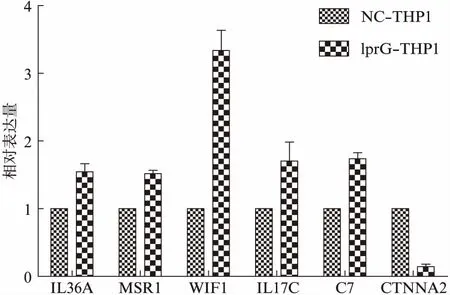
图5 通过qPCR方法验证RNA-seq的结果Fig.5 Results of real-time qPCR and RNA-seq
近年来,由慢病毒改建而来的慢病毒载体因其高效稳定的转染效率成为常用的研究工具,慢病毒载体可将外源基因有效地整合到宿主染色体上,长期稳定地表达目的基因[15]。本实验成功构建了过表达lprG的慢病毒载体,与包装载体共同包装成慢病毒,通过慢病毒介导的方法建立稳定表达lprG的THP1细胞系,进而利用转录组测序技术对稳定表达lprG的THP1细胞中的差异表达基因进行了筛选,结果显示有712个基因表达受到lprG过表达的影响。进行GO分类分析后发现差异表达基因主要分布在细胞内及膜表面,以蛋白质结合、催化活性及信号转导活性等功能为主,参与氨基酸及其衍生物代谢和细胞内处理等生物学过程。KEGG通路分析后显示差异基因主要富集在信号转导、免疫系统应答及感染性疾病途径中。免疫应答的启动及细胞内信号转导是宿主抵抗Mtb的重要机制,lprG的过表达影响免疫应答通路相关基因的差异表达,提示这些基因很可能是Mtb干扰宿主免疫应答的作用位点。
转录组测序结果表明lprG-THP1细胞中C7、IL17C和MSR1的表达均上调,该结果得到了qPCR的验证。研究[16]表明,C7、IL17C和MSR1在宿主抵抗Mtb的免疫应答过程方面发挥着关键的作用。巨噬细胞和树突状细胞可产生补体因子7(complement 7, C7),高水平的C7会减少细胞因子TNF-α和IFN-γ的分泌,加重小鼠肺内炎症及肝内的Mtb菌量。巨噬细胞清道夫受体1(macrophage scavenger receptor, MSR1)作为模式识别受体,结合病原体的配体成分以介导内吞作用,它通过调节吞噬作用影响免疫应答反应[17-18]。IL-17C是IL-17家族的一员,IL-17能通过影响P53途径抑制巨噬细胞的凋亡以促进Mtb的生长[19]。一方面,抑制巨噬细胞凋亡是Mtb长期寄居在巨噬细胞内的一种策略,另一方面,促进细胞凋亡有利于lprG感染邻近的巨噬细胞[20]。在本次实验过程中,未观察到lprG-THP1细胞凋亡的现象,与文献中所述lprG不会引起细胞凋亡一致[10]。目前,在国内lprG调控THP1细胞基因转录的实验尚未报导,lprG如何影响C7、IL17C和MSR1基因上调的具体机制也仍未明确,探讨相关信号通路机制将为发现治疗结核病的新型靶点提供参考。
Mtb合成前脂蛋白后会在信号肽的指导下完成翻译后脂肪修饰(如酰化)及转运,脂肪修饰方式利于脂蛋白锚定在细胞壁上,且脂蛋白N-端半胱氨酸残基的酰化使得lprG具有结合刺激TLR2的效能。当机体感染Mtb后,酰化的lprG会被巨噬细胞TLR识别后随Mtb吞噬入胞内,N-端半胱氨酸未酰化的lprG同样具有激活TLR2的活性[21]。本实验通过直接扩增Mtb的lprG基因序列,以慢病毒的方式在THP1细胞中表达内源性的lprG,由于THP1细胞不具有脂肪修饰lprG相关酶类,内源性与外源性的lprG在修饰方式及转运途径上也许会有不同,但也不能完全否定内源性表达的lprG不能激发相似的细胞效应。此外,本实验所用的THP1细胞为人单核源性巨噬细胞,此细胞模型只能模拟lprG与巨噬细胞的相互作用,并不能完全反映Mtb与人肺内巨噬细胞相互作用情况。本研究只是初步了解lprG对THP1细胞转录水平的影响,lprG与差异基因具体的相互作用机制将在后续实验中继续开展。
