Modulation of Molecular Sensing Properties of Graphdiyne Based on 3d Impurities
2018-09-07CHENXiZHANGShengli
CHEN Xi , ZHANG Shengli
1 Department of Applied Physics, School of Physics and Optoelectronic Engineering, Xidian University, Xi’an 710071, P. R. China.
2 Department of Applied Physics, School of Science, Xi’An Jiaotong University, Xi’an 710049, P. R. China.
3 MOE Key Laboratory for Non-equilibrium Synthesis and Modulation of Condensed Matter, Xi’an Jiaotong University, Xi’an 710049, P. R. China.
Abstract: In recent years, the successful preparation of single-layer graphene, MoS2, and other two-dimensional materials has started a new era of two-dimensional materials. The potential applications of twodimensional materials in emerging electronics have drawn widespread attention. Two-dimensional carbon materials, with their unique properties, have become the research hotspot of condensed matter physics, nanoelectronics, and biological medicine. The remarkable success in preparing graphene provides additional possibilities for developing sensitive biodevices and medicine systems. However, graphene is gapless and thus is unsuitable for building nanoelectronic devices or biosensors due to the too low on/off current ratio. More than 20 years ago, graphyne and its family (viz. graphdiyne, graphyne-3, etc.), as hypothetical C allotropes, were theoretically predicted to be semiconductors with a layered structure. Recently, graphdiyne was successfully synthesized on the surface of copper via a cross-coupling reaction using hexaethynylbenzene. Graphdiyne, as a new two-dimensional carbon material with semiconductor properties and a unique porous structure, is more advantageous than graphene for nanoelectronic and biosensing applications. As the first discovered semiconducting two-dimensional carbon material, with independent intellectual property rights in China,graphdiyne has great research significance. Compared with graphene, graphdiyne has a unique structure with larger pores composed of high π-conjugated acetylenic bonds, which may facilitate strong adsorption to biomolecules. Therefore,further research is needed to reveal how the physical properties of graphdiyne can be modulated effectively to meet the requirements of practical applications. The interaction between biological molecules and materials is an important subject of research in condensed matter physics and materials science. Detailed understanding of the interactions between graphdiyne and small molecules may facilitate the development of advanced biological applications such as biosensors for the detection of biomolecules and living cells, drug delivery systems, and cell imaging technologies. In sensitive analysis,the ultimate goal is to achieve reliable detection of trace amounts of molecules. In this work, first-principles calculations were employed to investigate the electronic structure of graphdiyne nanoribbons and the adsorption of graphdiyne to small molecules. To improve the chemical response of graphdiyne to single molecules, we considered modifying graphdiyne by doping 3d transition metal atoms. We chose Sc and Ti, which have the largest adsorption energies on graphdiyne, and studied the room-temperature stabilities of Sc- and Ti-doped graphdiyne and the possibility of using Sc- and Ti-doped graphdiyne as materials for molecular sensing. Finally, we investigated the interaction between graphdiyne and amino acid molecules and discovered that the dispersion force plays a large role in the interaction. The influence of amino acids on the electronic transport properties of graphdiyne was also studied, and the potential applications of graphdiyne to biosensors were investigated.
Key Words: Graphdiyne; Two-dimensional material; Electronic transport; Nanodevice
1 引言
碳元素是人类最早认识的元素之一,也是自然界成键结构最丰富的元素1。碳元素可构成许多具有特殊性质的材料2–6。在古代人们便发现了碳的同素异形体,如石墨和金刚石。随着近二十年来实验技术的发展,人们成功制备了许多不同结构的碳纳米结构,对材料技术的进步具有里程碑意义。含碳材料由于其具有的独特电子特性,在某些方面有许多先进应用,如太阳能电池,有机发光二极管,场效应管,化学传感器等。
2004年,Geim等人以单晶石墨为原料,得到了单原子层的石墨烯7,8。此后,人们发现了越来越多的二维材料。二维材料电子特性的潜在应用引起了广泛的关注。由于二维单原子层具有特殊的结构和独特的物理性质8–11,因此一直是众多领域中科学工作者研究的热点。然而,零带隙的石墨烯不适用于制备晶体管等电子器件。早在数十年前,就有人从理论上预测出一类由sp和sp2杂化的碳原子构成碳的同素异形体—石墨炔12,13。石墨炔具有平面层状结构,是由碳六元环和炔键构成的半导体。近年来,中国科学院化学研究所的李玉良院士课题组在实验上首次在铜表面成功制备出γ-石墨二炔(属于石墨炔中的一种,以下简称石墨二炔)14,15。石墨二炔是第一种具有半导体性质的碳的同素异形体,带隙与硅接近,比石墨烯更适用于制备纳米电子器件15–17,是新一代场效应管材料的候选者。
石墨二炔是我国独立研发并具有自主知识产权的新型二维碳材料。在技术发展迅猛的今天,传统电子学器件不断地小型化,在10 nm尺寸以下即将面受量子效应瓶颈。因此,人们迫切需要找到能取代传统硅基材料的新型纳米半导体材料。在纳米科技发展日新月异的今天,我们亟需发展基于石墨二炔的二维电子学及生物器件。其中所面临的主要问题有二:如何有效调控石墨二炔物理性质使其满足实际应用的需求;如何根据石墨二炔的特性提出电子学器件的设计方案。石墨二炔具有独特的由 π共轭炔键构成的大孔网状结构,可被应用于气体分离膜、储能材料、电池电极材料、太阳能电池材料18–20等领域。此外,石墨二炔亦对生物分子有吸附作用。基于石墨二炔的独特优势,我们考虑石墨二炔将应用到生物器件领域。由于氨基酸是生命体基本单元,我们需掌握氨基酸及其它生物小分子与石墨二炔表面相互作用规律及其物理机制,这是设计基于石墨二炔的纳米生物传感器的基础。
分子检测的理想目标为达到终极的检测灵敏度,即能够对单个分子进行解析。迄今为止,人们已经发展了大量的检测技术手段,如扫描隧道显微镜、原子力显微镜21和荧光关联光谱22等等。现代的扫描隧道显微镜技术,已能够在单分子水平上操作化学合成23。荧光关联光谱主要用于检测聚合物折叠24、大分子构象的改变25及较快的酶动力学过程26,27。现今,人们更多地把注意力集中在小尺寸的分子检测器上,并要求具备高灵敏度及易用性。二维材料的诞生给气体检测器的制备开拓了更多的可能性。二维材料具有非常大的比表面积,高的灵敏度。气体分子在二维材料表面吸附后,起到了电子施主或受主的作用,能引起二维材料载流子浓度的改变,从而形成电信号响应。人们已经利用石墨烯的霍尔效应来检测吸附气体分子造成的载流子浓度改变28,并解析出单个分子的信号。基于石墨烯的场效应管也开发并用于检测单分子的电信号29–32。为了提高检测的灵敏度,人们考虑各种对二维材料表面进行修饰的方法。过渡金属原子往往可作为小分子的吸附中心。而石墨烯表面的π键是饱和的,具有化学惰性;因此金属原子在石墨烯表面的吸附能低于2 eV,迁移势垒低于0.8 eV33,34。较低的吸附能和迁移势垒将使得金属原子在室温下聚集。相比之下,石墨二炔比石墨烯更容易吸附金属原子,使之成为潜在的分子吸附材料。这是石墨二炔在分子检测材料中的优势。
在本文中,我们首先研究了石墨二炔纳米带的电子输运性质。之后,考虑利用石墨二炔的特殊大孔碳网状结构作为小分子传感器材料。为了提高石墨二炔的响应,在3d过渡金属中,我们挑选出了在石墨二炔表面吸附能最大的Sc、Ti原子,并确定了Sc、Ti掺杂石墨二炔在室温下的稳定性。我们选择HCHO作为典型的有代表性的小分子,从能带、载流子浓度等方面探讨了Sc、Ti掺杂石墨二炔对HCHO分子的响应。我们还研究了氨基酸与石墨二炔的相互作用及物理机制,得出氨基酸对石墨二炔光吸收和电输运性质影响的规律。本研究为今后将石墨二炔用于新型生物分子电子学及生物学检测器件,在生物电子学及药物载体方面的应用提供重要理论指导。
2 理论模型与计算方法
在本文中,我们主要使用SIESTA软件包35,采用密度泛函理论计算石墨二炔的电子性质。在计算中采用Perdew-Burke-Ernzerhof (PBE)36的广义梯度近似(GGA)方法来描写电子的交换关联作用,引入了包含色散力的PBE-D2修正37,以及模守恒的Troullier-Martins赝势38。实空间积分格点截断取为250 Ry。使用了极化的双ζ基组对波函数进行展开。对二维的石墨二炔单层,沿与其平面垂直的方向加上了2.0 nm的真空层。进行计算前,我们先将各种结构进行驰豫,直到所有原子受力皆小于0.1 eV·nm−1。在分子动力学模拟中,我们使用了Verlet算法对原子的牛顿方程进行积分,时间步长为1 fs,采用Nose热浴控制体系温度。
考虑到PBE泛函往往低估带隙,因此我们在能带计算中采用杂化的 Heyd-Scuseria-Ernzerhof(HSE06)泛函39,40,配合 VASP 软件41–43在 PAW 方法下进行计算。平面波能量截断为400 eV。在2 ×2 × 1的Γ中心Monkhorst-Pack采样下选取k点进行自洽计算,而后在布里渊区高对称路径上取30个k点计算能带。
在介观输运计算中,为了缩短计算时间,我们使用了极化的单ζ基组。纳米带k点用1 × 1 × 100进行采样。在系统两端施加偏压后,用非平衡格林函数方法44(TRANSIESTA软件包45)计算电子的透射率,然后根据 Landauer-Buttiker公式计算电流。
3 石墨二炔纳米带电子结构与电子输运性质

图1 (a) 2 × 2的石墨二炔原子结构,其中虚线范围表示原胞;(b) PBE和HSE06泛函给出的2 × 2石墨二炔能带(左)和态密度(右)Fig. 1 (a) The structure of graphdiyne 2 × 2 supercell, primitive cell is denoted by dashed lines;
石墨二炔是由sp杂化的碳原子链与sp2杂化的碳六圆环组成的二维平面碳网。从图1a中我们看出,石墨二炔为二维平面网络状的碳结构,其中碳碳三键(sp杂化)是构成石墨二炔结构中十分重要的连接单元。由于它不会受顺反异构的变化影响,因此可以一直保持线性的结构。其次,它具有更小的空间位阻,这很有利于将sp杂化的碳连接到sp2杂化或者sp杂化的碳原子中心上。另外,碳碳三键与苯环之间形成了离域π键,因此其高度共轭、碳富集的有机分子结构具有优良的灵活可调性。虽然石墨烯具有比硅更快的电子移动速率,但是石墨烯并不具有明显的能带隙,不适合被应用于场应效晶体管。而石墨二炔具有直接带隙,其值约为1 eV。相比石墨烯,石墨二炔更适合被应用于场效应晶体管。值得注意的是,石墨二炔的带隙值与硅的(1.1 eV)很相近。由于以硅为基础的科学技术拥有其自身的局限性,石墨二炔将有可能代替硅来制成更快更小的电子学器件。而且石墨二炔直接带隙的存在,促进了光—电的高效转换,有助于其在光电学器件上的应用。这些优点就使得石墨二炔很可能替代硅成为电子学器件的优异材料。
石墨二炔的原胞如图1a中的菱形虚线所示,菱形的边长即为石墨二炔的晶格常数a0。我们的计算得到a0= 0.950 nm,与文献46的计算值a0=0.948 nm吻合。能带计算表明,石墨二炔具有直接带隙。在后续的计算中(见第4、5节),为了研究系小分子吸附,我们取了2 × 2晶胞作为衬底模型进行能带计算。图1b左给出了PBE和HSE06泛函所计算得到的能带及态密度。我们通过PBE泛函计算出的带隙为0.44 eV,与文献值0.46 eV46相近。HSE06泛函计算出的带隙为0.83 eV,与文献值(0.88 eV)47接近。这里需指出的是,普通的密度泛函方法计算的带隙总是偏小,PBE计算值低于带隙实验值。在文献47中,准粒子(GW)准粒子近似给出的带隙1.10 eV最接近实验值。GW准粒子近似对带隙的修正将近1.5倍,这是由低维材料中较强的电子间库伦相互作用所引起。图1b右展示了PBE和HSE06泛函所给出的石墨二炔态密度。
为了研究石墨二炔作为介观器件材料的电子输运特性,我们考虑以平直的石墨二炔纳米带作为理论研究模型。在进行计算之前,先对石墨二炔纳米带的结构进行分类。我们考虑了扶手椅型(AGDNR)和锯齿型石墨二炔纳米带(ZGDNR)。各种结构、各种宽度和各种边缘形状的石墨二炔纳米带结构如图2a所示。n-AGDNR边缘平直,带有六元环。这里的整数n表示其相对宽度(横向碳原子排数)。ZGDNR边缘为突起状,类似于锯齿型石墨烯纳米带。但ZGDNR突起比锯齿型石墨烯纳米带大得多。ZGDNR分为两类,其中n-ZGDNR两边缘是对称的,而n’-ZGDNR两边缘的锯齿位置是错开的。纳米带沿z方向具有周期性,我们选择基本重复单元(图2a黑线框)作为原胞进行几何结构优化。计算结果表明,图2a中各种纳米带的碳六圆环间距a与石墨二炔晶格常数a0差别在2%以内,这说明石墨二炔的纳米带结构与层状石墨二炔接近。
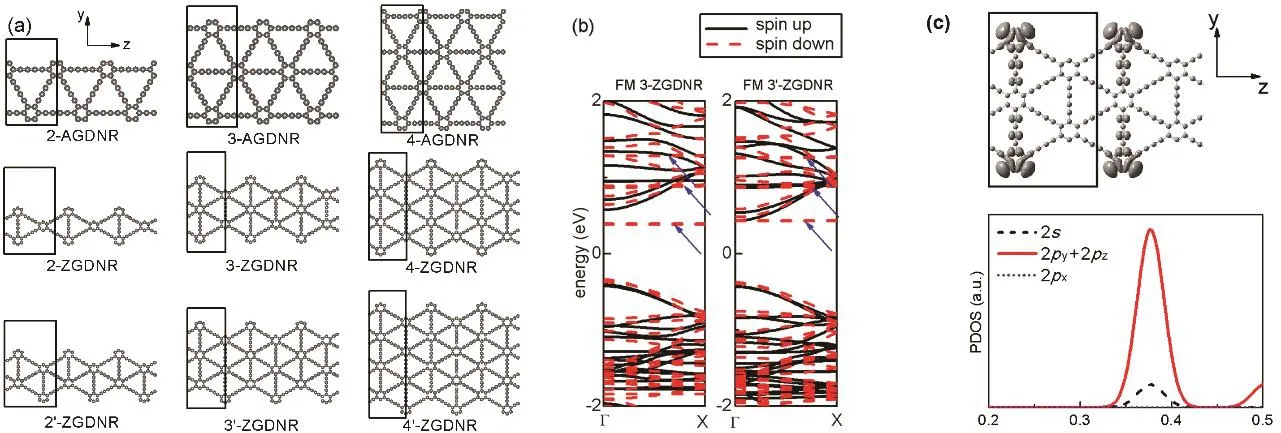
图2 (a)不同宽度的扶手椅型石墨二炔纳米带和两种类型的锯齿型石墨二炔纳米带,分别表示为n-AGDNR、n-ZGDNR及n’-ZGDNR,其中黑线框标出每条纳米带的晶胞;(b)铁磁态(FM)的3-ZGDNR与3’-ZGDNR的能带图,边缘态能带用蓝色箭头标出;(c)铁磁态(FM) 3-ZGDNR的电荷密度图及其边缘态轨道态密度(PDOS)图Fig. 2 (a) Armchair and two different types of graphdiyne nanoribbons with different widths, which are called
从能带结构可以看出,各种不同宽度的石墨二炔纳米带均为直接带隙半导体,带隙出现在Γ点。此处为了节省计算时间,用PBE泛函计算带隙,仅用于定性比较。2-,3-,4-和5-AGDNR的带隙分别为0.79,0.70,0.65和0.61 eV。由于量子限域效应,石墨二炔纳米带宽度越小,带隙越大。随着石墨二炔纳米带宽度的增大,带隙下降并越来越接近无限大石墨二炔平面的PBE带隙值(0.47 eV)。n-ZGDNR (表1)和n’-ZGDNR (表2)的带隙亦表现出此特性。
我们考虑了石墨二炔纳米带的各种磁性状态。计算结果表明,各种AGDNR均不具有磁性。而 ZGDNR具有磁性,其铁磁(FM)态能量比相应的非磁态(NM)能量低(表 1和表 2分别给出了 n-ZGDNR和n’-ZGDNR的带隙)。FM态的磁矩主要分布在 ZGDNR边缘突起部分的原子上。对各种不同形状、不同宽度的n-ZGDNR,FM态比NM态的每晶胞能量低1.01–2.12 eV。对n’-ZGDNR,FM态比NM态的每晶胞能量低2.00–2.04 eV。此外,我们还考虑了两种反铁磁(AFM)态。第一种情况为,相邻ZGDNR晶胞磁矩反平行排列(AFM1)。结果表明对同种ZGDNR,AFM1态的能量比FM态高约0.01–0.05 eV。第二种情况为,纳米带两边缘磁矩反平行排列的情形(AFM2)。结果表明对同种ZGDNR,AFM2态的能量比同种结构的FM态高约0.08–0.10 eV。综上所述,FM态为ZGDNR的最稳定状态。

表1 n-ZGDNR的带隙(PBE泛函)、FM态与NM态能量差(EFM − ENM)、FM态晶胞磁矩(μ)Table 1 Band gap, total energy difference between FM and NM state (EFM − ENM) and the magnetic moment of FM unit cell (μ) of n-ZGDNRs at the level of PBE.

表2 n’-ZGDNR的带隙(PBE)、FM态与NM态能量差(EFM − ENM)、FM态晶胞磁矩(μ)Table 2 Band gap, total energy difference between FM and NM state (EFM − ENM) and the magnetic moment of FM unit cell (μ) of n’-ZGDNRs.
对各种形状、各种宽度的ZGDNR,由于其铁磁基态特性,能带均表现出自旋极化特征。作为例子,图2b显示了FM态的3-ZGDNR和3’-ZGDNR的能带。在能带图中,能够明显看到若干特殊的呈现出几乎平直E–k关系的能带(蓝色箭头)。我们猜测这些电子态为局域电子轨道。为了揭示这些平直能带的本质,我们计算了它们的电荷密度分布,及其轨道投影态密度(PDOS) (图2c)。从电荷分布可以看出,其电子云主要局域在锯齿型边缘的突起处,表明这些局域态为纳米带的边缘态。由于ZGDNR的边缘突起彼此间隔距离较远(比锯齿型石墨烯边缘突起间距大),各个锯齿尖端的边缘态的相互作用较弱。所以这些局域态相对独立,形成了这些平直带。从其 PDOS可以看出,边缘态主要由2s轨道及面内2py+ 2pz轨道组成。
为了研究石墨二炔纳米带的介观电子输运性质,我们构造了图3a所示的模型。以纳米带为两端电极(模拟时取原胞加上周期边界条件)、3倍原胞长度作为散射区进行计算。在两端施加电压 Vb后,计算通过体系的电流I。我们也尝试过选取更长的散射区,结果 I–Vb曲线无明显变化,因此 3倍原胞长度的散射区是足够大的。对各种纳米带,I–Vb曲线呈现出半导体特征。图 3b展示了 2–4-AGDNR的I–Vb曲线。当eVb大于带隙时电流I为几到十几μA量级,当eVb小于带隙时电流I极小(小于0.01 μA)。从图3b可以看出,AGDNR的宽度越大,带隙越小,导通电压越小。当eVb大于带隙时,AGDNR导通,I–Vb曲线近似为直线。在导通区域,2–4-AGDNR的电导约为0.77–1.31 G0。4-AGDNR的电导最大。对各种 n-ZGDNR和 n’-ZGDNR,I–Vb曲线也呈现出半导体特征。图3c呈现了 n-ZGDNR 的 I–Vb曲线。图 3d呈现了 n’-ZGDNR的 I–Vb曲线。这些曲线均具备半导体特征,与 AGDNR类似。总的来看,纳米带宽度越宽,导通后的导电能力越强。对 3-ZGDNR、3’-ZGDNR、4-ZGDNR、4’-ZGDNR,导通段的电导约为0.26–0.60 G0。值得一提的是,FM的ZGDNR产生自旋极化电流。自旋极化率定义为P = | I↑−I↓|/(I↑+ I↓),其中 I↑和 I↓分别为自旋向上和自旋向下电流。在导通段,3-ZGDNR、3’-ZGDNR、4-GDNR和 4’-ZGDNR的电流自旋极化率为 23%–64%。
4 Sc、Ti掺杂石墨二炔对HCHO分子的吸附与响应
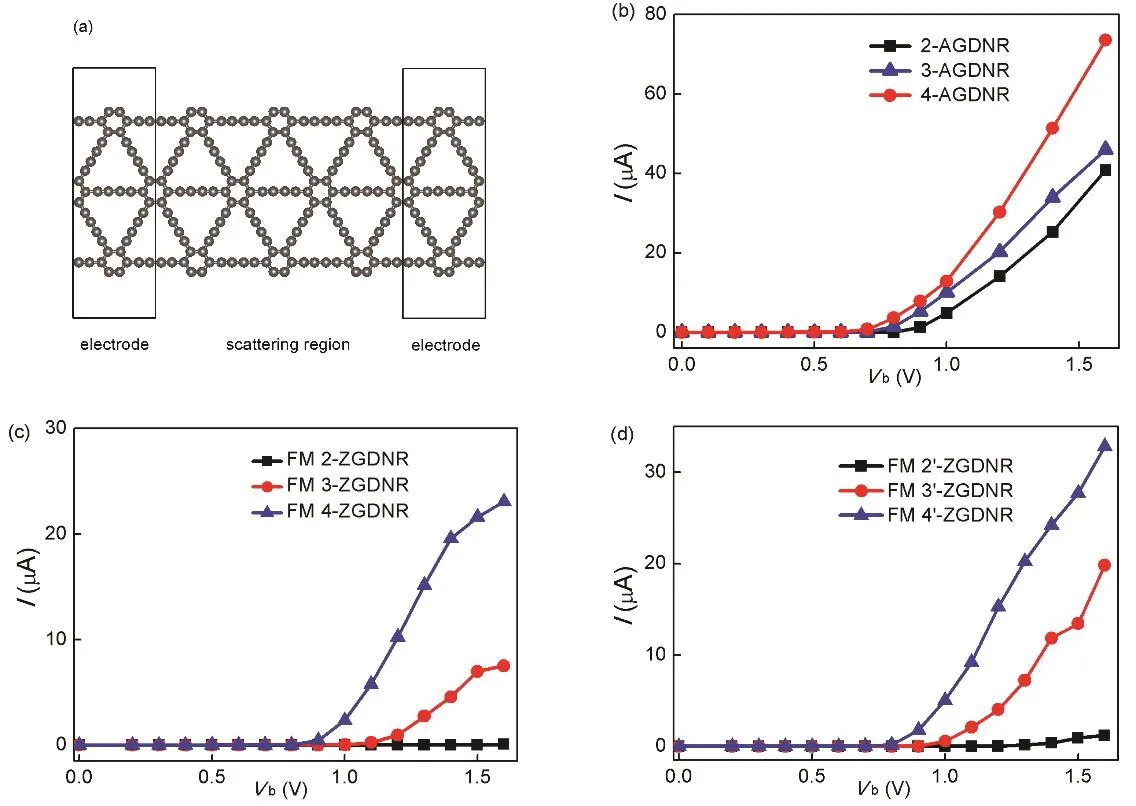
图3 (a)石墨二炔纳米带介观输运计算模型,(b) 2–4-AGDNR的I–Vb曲线,(c) 2–4-ZGDNR的I–Vb 曲线,(d) 2’–4’-ZGDNR 的 I–Vb 曲线Fig. 3 (a) The calculation model of electronic transport in graphdiyne nanoribbons, (b) I–Vb curves of 2–4-AGDNRs,(c) I–Vb curves of 2–4-ZGDNRs, (d) I–Vb curves of 2’–4’-ZGDNRs.
接下来,我们考虑将石墨二炔作为小分子传感器的可能性。欲作为传感器材料,首先要求此种材料对小分子具有一定的吸附能力。目前的研究表明,石墨炔的饱和炔键结构对一些小分子(如H2、O2、CO、N2、CH4)的吸附较弱48,49,其相互作用主要是微弱的排斥作用。为了提高石墨二炔的吸附能力,我们考虑在其大孔中加入3d金属原子。
这里选择HCHO分子作为典型的研究对象。为了计算HCHO分子在石墨二炔上的吸附,我们选取各种不同初始位置及不同的HCHO分子取向来进行结构优化,寻找最稳定的吸附构型(即吸附能最大的构型)。模拟中采用2 × 2的石墨二炔晶胞,在2 × 2 × 1的Γ中心Monkhorst-Pack采样下选取k点进行自洽计算。结果表明,当HCHO分子位于三角形炔链中心处(图 4a)时为在石墨二炔表面最稳定的吸附构型,吸附能为 Ead= 0.43 eV(表3)。此外,若HCHO分子位于石墨二炔的碳六圆环上,则吸附能为负值,为不稳定位置。相比之下,我们也获得了HCHO在石墨烯表面的最稳定吸附位置,相应的最大吸附能是Ead= 0.28 eV,明显小于在石墨二炔表面的吸附能。作为参考,我们以相同的方法也搜索了CO和C6H6分子在石墨二炔和石墨烯上的最稳定吸附构型。CO分子在石墨二炔和石墨烯上的最大吸附能Ead(CO)分别是0.55和0.28 eV,C6H6分子在石墨二炔和石墨烯上的最大吸附能Ead(C6H6)分别是0.81和0.75 eV (表3)。结果表明,两种分子在石墨二炔上的吸附能都比在石墨烯上大。与石墨烯中的烯键相比,石墨二炔中炔链中额外的π电子可能会导致吸附增强。
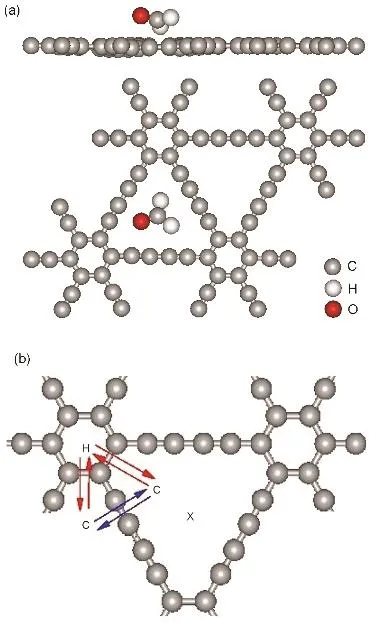
图4 (a) HCHO分子在石墨二炔上吸附的侧视及俯视图;(b) C、H、X表示Sc、Ti原子三种可能的吸附位置Fig. 4 (a) Side and top views of HCHO adsorption on graphdiyne; (b) three possible adsorption sites of Sc and Ti atoms shown by C, H and X.

表3 HCHO、CO、C6H6分子在石墨二炔和石墨烯上的吸附能EadTable 3 The adsorption energy Ead of HCHO,CO and C6H6 molecule on graphdiyne and graphene.
为了将3d金属原子掺杂的石墨二炔作为分子传感材料,我们首先要确定其在室温下的稳定性。对3d金属原子,我们考虑了在石墨二炔表面三种可能的吸附位置,如图4b所示。分别为由炔链组成的三角形大孔中心X位置、大孔角上C位置和六圆环中心H位置。几何优化结果表明,C位置和H位置是稳定的吸附位置。而X位置不稳定,因为在优化过程中,Sc、Ti原子会自发迁移至 C位置。对于ScTi原子在C位置的吸附能Ead(C)大于H位置的吸附能Ead(H),如表4所示。我们也考虑了其他的3d过渡金属原子的吸附,发现Sc、Ti原子在石墨二炔表面的吸附能大于或接近 V、Cr、Mn,Fe、Co、Ni吸附原子在石墨二炔表面的吸附能50。从稳定性角度看,Sc、Ti原子是3d过渡金属元素中最好的选择。值得一提的是,Sc、Ti吸附原子在石墨二炔表面的吸附能,比在石墨烯表面吸附能(Ead< 2 eV)33,34,51大得多。因此,石墨二炔比石墨烯更适宜做过渡金属原子的基底。
接下来,为了解Sc、Ti原子在石墨二炔表面的热稳定性,我们计算了它们在石墨二炔表面迁移的最小能量路径以及势垒E0。如图4b中红箭头所示,Sc、Ti原子可从C位置,先跳到最近邻的H位置再迁移至另一个C位置。也可以如图4b中蓝箭头所示,越过炔链直接迁移至相邻的C位置。根据计算结果,C→H→C的势垒比C→C势垒大两倍,因此越过炔链的C→C迁移是Sc、Ti原子迁移的最小能量路径。此外,Sc、Ti原子也可能会在炔链三角形大孔内迁移。如果越过炔链的C→C势垒足够大,Sc、Ti原子只能停留在同一个炔链三角形内部。计算得到的 Sc和 Ti原子的C→C扩散势垒 E0分别为 2.19和 1.74 eV (见表4)。一般来说,室温下扩散势垒大于0.8 eV的反应是难以进行的。也就是说,在石墨二炔表面,足够高的C→C扩散势垒能阻止Sc、Ti原子聚集。所以我们可以推断,在室温下Sc、Ti原子能很好地稳定分散在石墨二炔表面。

表4 Sc、Ti原子分别在石墨二炔的C位置吸附能Ead(C)和H位置的吸附能Ead(H),在C与C位置间的扩散势垒E0Table 4 The adsorption energy of Sc and Ti atoms on the C and H sites of graphdiyne (Ead(C) and Ead(H),respectively), and the migration barrier E0 between neighboring C sites.
研究HCHO在Sc、Ti掺杂石墨二炔上的吸附情况,是了解其作为分子传感器的先决条件。我们随机选取HCHO在掺Sc石墨二炔和掺Ti石墨二炔表面上各种初始的几何构型,通过几何优化来找到最稳定的吸附构型。石墨二炔-Sc和石墨二炔-Ti都表现出与HCHO分子的较强的相互作用。图5展示出石墨二炔-Sc·HCHO和石墨二炔-Ti·HCHO 的最稳定吸附构型。在石墨二炔-Sc·HCHO构型中,HCHO分子中的O原子与 Sc原子成键。在石墨二炔-Ti·HCHO构型中,HCHO中的C原子和O原子同时与Ti原子成键。HCHO在石墨二炔-Sc和石墨二炔-Ti上最稳定构型的吸附能分别是2.59和2.24 eV,远大于在纯石墨二炔上的吸附能 0.43 eV。相比之下,我们还研究了HCHO分子在石墨二炔-K、石墨二炔-Ca和石墨二炔-Cr体系上的吸附情况,吸附能分别是1.66、1.60和1.58 eV,均低于在石墨二炔-Sc和石墨二炔-Ti上的吸附能。相比之下,HCHO分子在掺杂Sc和Ti的石墨二炔上的吸附相当强。此外,我们还研究了多个HCHO分子在石墨二炔-Sc和石墨二炔-Ti上的吸附。几何优化说明,在吸附一个HCHO分子后石墨二炔-Sc·HCHO 和石墨二炔-Ti·HCHO体系对于额外HCHO分子产生排斥效应。从能量角度看,以上结果表示Sc、Ti吸附原子只能结合一个HCHO分子。这个特点为单分子的传感提供了前提条件。

图5 HCHO分子在石墨二炔-Sc和石墨二炔-Ti上吸附的侧视及俯视图Fig. 5 The side and top views of HCHO adsorption of graphdiyne-Sc and graphdiyne-Ti systems.
为了研究石墨二炔-Sc和石墨二炔-Ti体系对HCHO分子探测的灵敏度,我们比较这两个体系了吸附HCHO分子前后,两个体系的载流子浓度。在第一性原理计算中,导带中的电子浓度n为

其中,能量 E的积分下限为导带底(CBM),D(E)为态密度,EF为费米能量。价带中的空穴浓度 p为
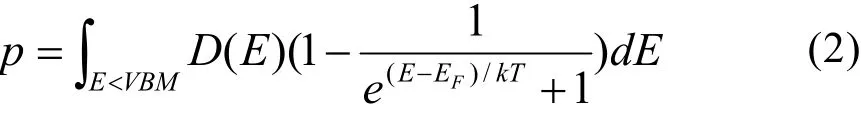
其中,能量 E的积分上限为价带顶(VBM)。总载流子浓度N = n + p。为了较准确地估算载流子浓度,各个体系的能带均由杂化泛函HSE06的计算得到,如图6所示。通常,PBE泛函的计算结果会低估半导体能带,由杂化泛函HSE06计算的结果与PBE相比更精确。PBE泛函和HSE06泛函的计算结果见表5。
对于纯净石墨二炔的2 × 2超胞,能带表现出本征半导体特性(即n = p),由PBE和HSE06计算得到的Γ点直接带隙分别为Eg= 0.48和0.89 eV。室温300 K下由PBE和HSE06计算得到相应的载流子浓度为 N = 3.96 × 108,1.29 × 105cm−2(见表5)。吸附HCHO分子后,体系带隙略微减小到Eg= 0.46/0.88 eV (PBE/HSE06),室温下相应的载流 子 浓 度 N = 4.71 × 108/1.47 × 105cm−2(PBE/HSE06)比吸附前的纯净石墨二炔高16%/12%,增加的载流子浓度N主要是由于HCHO分子导致的带隙减小。
在石墨二炔-Sc及石墨二炔-Ti体系中,Sc、Ti吸附原子为石墨二炔贡献了额外的电子,使导带移至费米面下使石墨二炔转变成导体(图6b、c)。室温300 K下,石墨二炔-Sc体系载流子浓度为N =1.21 × 1013/1.30 × 1013cm−2(PBE/HSE06),石墨二炔-Ti体系载流子浓度为 N=1.06 × 1013/1.23 × 1013cm−2(PBE/HSE06)。我们选取的 Sc、Ti与 2 × 2石墨二炔超胞体系的掺杂浓度c = 3.11 × 1013cm−2,相当于每个 Sc原子对石墨二炔贡献了 0.42个电子,而每个Ti原子对石墨二炔贡献了0.4个电子。电荷分析表明,石墨二炔上的 Sc原子及 Ti原子的净电荷分别为+0.74e和+0.55e。以上结果展示了石墨二炔上的Sc、Ti吸附原子对电荷的贡献。石墨二炔-Sc及石墨二炔-Ti体系比纯石墨二炔体系载流子浓度大得多。
吸附一个HCHO分子后,石墨二炔-Sc·HCHO与石墨二炔-Ti·HCHO 体系中的导带都相对石墨二炔-Sc及石墨二炔-Ti中的向上移动(图6b、c)。这一结果展示了HCHO分子的电负性。HCHO分子从石墨二炔中吸引电子,降低载流子浓度。当温度为300 K,石墨二炔-Sc·HCHO体系呈现出金属性,载流子浓度N = 1.28 × 1013cm−2(HSE06泛函的结果),略小于石墨二炔-Sc的(见表5)。对于石墨二炔-Ti·HCHO体系,导带甚至移到费米面上,体系由金属性转变为n型半导体特性。在室温300 K下,其载流子浓度N = 6.19 ×106cm−2(HSE06泛函的结果),比石墨二炔-Ti体系低几个量级,见图6c。这表面HCHO分子几乎全部带走了Ti原子对石墨二炔的电子贡献。所以,石墨二炔-Ti对于吸附 HCHO分子的电子响应比石墨二炔-Sc更敏感且强得多。
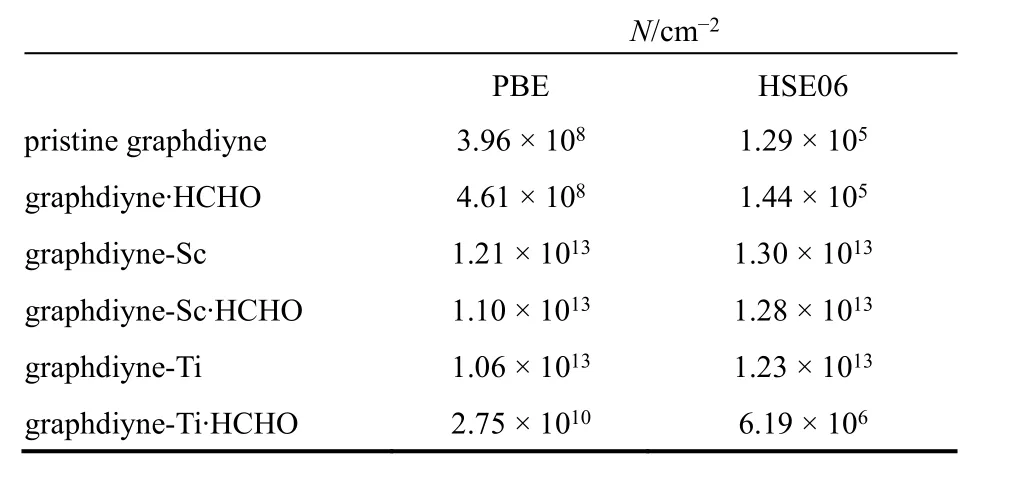
表5 由PBE和HSE06两种泛函计算得到的石墨二炔相关体系的载流子浓度N = n + p。Table 5 Carrier concentration N = n + p of graphdiyne systems at the level of PBE and HSE06.

图6 采用HSE06泛函计算得到的纯净石墨二炔2 × 2超胞、石墨二炔-Sc、石墨二炔-Ti在吸附HCHO 前后的能带图Fig. 6 The energy bands of pristine 2 × 2 graphdiyne, graphdiyne-Sc and graphdiyne-Ti at the level of HSE06.
5 石墨二炔与氨基酸的相互作用
为了研究石墨二炔与氨基酸分子的相互作用,考虑到氨基酸的R基侧链分为疏水、亲水两类。此外,氨基酸也按极性、非极性分类。在这里,我们选取甘氨酸(Gly)、谷氨酸(Glu)、苯丙氨酸(Phe)和组氨酸(His)作为每类氨基酸的代表。从氨基酸 R基侧链的极性来看,Phe是非极性不带电具有疏水性,Gly是极性不带电具有亲水性,His是极性带正电具有亲水性,Glu是极性带负电具有亲水性。从化学结构来看,Gly及Glu为脂肪族氨基酸,Phe为芳香族氨基酸,His为杂环族氨基酸。所选氨基酸优化后的结构如图7a所示。
在计算中,我们仍然采用2 × 2的石墨二炔晶胞作为衬底模型。此外,为了与氨基酸在石墨烯上的吸附进行比较,我们还将同样的模拟方法用于7 × 7的石墨烯晶胞(边长1.72 nm)。对石墨二炔和石墨烯单层,加上了2.0 nm的真空层。所选择的2 × 2的石墨二炔晶胞和7 × 7的石墨烯晶胞的尺寸,能够保证周期性盒子之间氨基酸分子的间距大于 0.9 nm。这样我们可以认为氨基酸分子是近独立的。为了测试极化双ζ基组的可靠性,我们以石墨烯为测试对象,用极化单ζ、极化双ζ和极化叁 ζ基组进行了结构优化,得到的碳碳键长分别为0.144、0.142和0.142 nm。可以看出,极化双ζ和极化叁 ζ基组已经足够合适。为了节省计算时间,在计算中我们采用的是极化双ζ基组。为了研究范德华力的作用,我们还使用了vdW-DF52,53泛函与PBE-D2泛函进行比较。
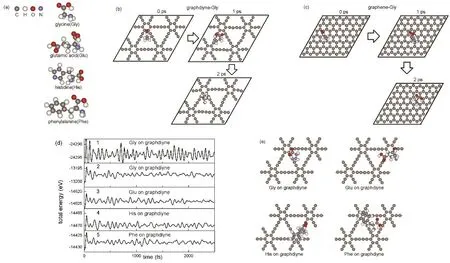
图7 (a)计算中涉及的四种氨基酸分子,甘氨酸(Gly)、谷氨酸(Glu)、苯丙氨酸(Phe)和组氨酸(His);(b)室温下Gly分子在石墨二炔表面的迁移;(c)室温下Gly分子在石墨烯表面的迁移;(d)分子动力学模拟中的总能量变化;(e) Gly、Glu、His和Phe在石墨二炔表面的最稳定吸附位置Fig. 7 (a) The structure of glycine (Gly), glutamic acid (Glu), histidine (His) and phenylalanine (Phe) molecules;(b) The migration of Gly on graphdiyne surface at room temperature; (c) The migration of Gly on graphene surface at room temperature; (d) The change of total energy in molecular dynamics simulations; (e) The most stable adsorption configurations of Gly, Glu, His and Phe on graphdiyne surface.
氨基酸在材料表面的吸附主要是由于范德华力的作用。范德华力分为取向力、诱导力和色散力三类。其中,取向力为极性分子与极性分子间的静电力。诱导力为极性分子靠近非极性分子后,将非极性分子极化后产生的静电力。色散力是一种特殊的力,它由偶极矩的量子涨落引起。一般的密度泛函方法(例如 PBE泛函36)不能很好的描述色散力。当分子距离较远时色散力占主要地位,若泛函不能描写量子涨落,计算出的分子间作用力会过快地趋于零。为了解决这个问题,人们发明了修正的非局域 vdW 泛函(如这里所使用的 vdW-DF),但这种泛函的计算量比一般的 GGA泛函大。此外,近年来流行的 DFT-D方法(如本文中使用的PBE-D2),将一般GGA泛函与经验势相结合,也是一种好的方案。下面我们采用PBE、vdW-DF泛函和 PBE-D2泛函计算氨基酸在石墨二炔表面的吸附能,并进行比较,揭示相互作用力的本质。
首先,我们以Gly为例,研究Gly分子与石墨二炔的相互作用,并与石墨烯进行比较。我们采用了分子动力学(MD)模拟的方法进行了初探。初始时,我们将一个位置及取向随机的 Gly分子置于石墨二炔表面进行结构优化,然后在恒温300 K下进行了分子动力学模拟。在模拟过程中,我们抽选某些体系势能较低的瞬态进行结构优化。从优化后的几何结构中找出势能最低,也就是 Gly吸附在石墨二炔表面的稳定结构。根据结果,最稳定的石墨二炔吸附Gly复合体系如图7e所示。在上述分子动力学模拟中,我们观察到在300 K温度下,Gly分子持续在石墨二炔表面迁移并转动。一些结构在展示与图 7b。在室温下,Gly分子迁移过程中与石墨二炔表面总是保持一定距离,但不会离开石墨二炔表面。在模拟中,总能量随时间的波动并保持在一定范围内(图7d2),该结果反映出体系处于平衡态。我们也进行了800 K下的分子动力学模拟,Gly分子仍然束缚于石墨二炔表面。这一结果证明 Gly在石墨二炔表面的吸附是稳定的。
为了研究Gly在石墨二炔表面的最稳定吸附,我们选取MD模拟过程中势能较低的几个构型进行优化。对PBE、PBE-D2和vdW-DF泛函,所得到的Gly最大吸附能分别为0.59、0.90和1.10 eV(表6)。计算结果表明,PBE泛函给出的吸附能明显地小于PBE-D2或vdW-DF泛函,这说明色散力在氨基酸吸附中占了很大一部分比例。PBE泛函对色散力的描写并不太好,不能表示长程-C6/R6衰减(R为分子间距)。而PBE-D2与vdW-DF泛函结果接近,说明这里的计算结果较为可靠。对于各个不同的吸附位,PBE-D2泛函给出的Gly与石墨二炔的吸附能范围是0.68–0.90 eV。由于Gly在石墨二炔表面的吸附构型较为复杂,模拟中很难遍历所有结构,故以上吸附能是大致的估计。在最稳定的吸附构型(图7e)中,Gly分子在石墨二炔表面位于大的碳环附近顶点。在 Gly分子中,由于 O原子与N原子具有强的负电性,O与N原子附近电子密度较大。当 Gly分子接近时,石墨二炔表面将被O或N原子极化,产生诱导力而被吸引。根据以上得到的Gly吸附构型,―COOH与―NH2同时靠近石墨二炔表面时具有最大的吸附能(图7e)。对于仅―COOH或―NH2一个靠近石墨二炔面的构型,吸附能则较低。

表6 采用PBE、PBE-D2和vdW-DF三种不同的泛函得到的Gly、Glu、His和Phe在石墨二炔表面的吸附能(单位:eV)Table 6 The adsorption energy (in eV) of Gly, Glu, His and Phe on graphdiyne surface at the level of PBE,PBE-D2 and vdW-DF.
为了与石墨烯进行比较,研究 Gly在石墨烯表面的吸附。我们采用了与石墨二炔吸附 Gly类似的方法进行了模拟。模拟的情况与石墨炔类似,在模拟中总能量随时间的波动并保持在一定范围内(图7d1),结果反映出体系处于平衡态。图7c展示了Gly在石墨烯表面运动的过程。对PBE、PBED2和vdW-DF泛函,所得到的Gly最大吸附能分别为0.23、0.53和0.54 eV (表7)。结果同样表明,PBE泛函给出的吸附能明显地小于 PBE-D2或vdW-DF泛函,色散力在氨基酸吸附中占了很大一部分比例。对PBE泛函,所得到的Gly在石墨烯表面最稳定构型的吸附能与文献中的计算结果54一致。最稳定的吸附构型是N、O、C原子所在平面与石墨烯面平行的状态。根据以上结果,Gly与石墨二炔的吸附明显强于与石墨烯的吸附。与石墨烯中烯键相比,石墨二炔中的炔键具有额外的π电子可能是导致与氨基酸分子的强吸引。
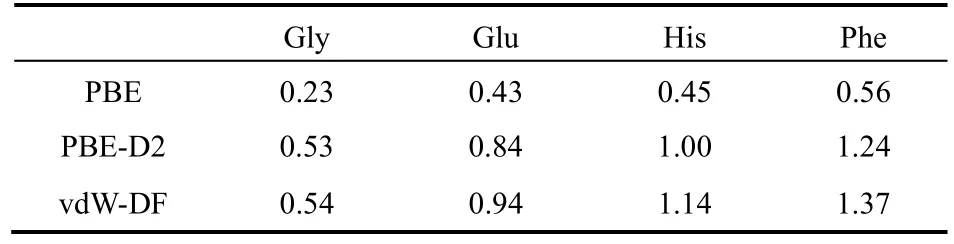
表7 采用PBE、PBE-D2和vdW-DF三种不同的泛函得到的Gly、Glu、His和Phe在石墨烯表面的吸附能(单位:eV)Table 7 The adsorption energy (in eV) of Gly, Glu, His and Phe on graphene surface at the level of PBE,PBE-D2 and vdW-DF.
我们用同样的方法研究Glu、His及Phe在石墨二炔表面的吸附。模拟温度为300 K。从任意的Glu、His和Phe的构型出发,从模拟中找出Glu、His和Phe势能较低的瞬态构型并进行结构优化。从中找出吸附能最大的构型。与Gly类似,在300 K的模拟中,Glu、His及Phe分子在石墨二炔表面持续迁移并转动,与表面保持0.2–0.3 nm的距离,但不离开石墨二炔表面。在模拟中,总能量随时间在一定范围内波动(图7d3、d4、d5分别表示Glu、His和Phe的情况)。这种波动反映出体系已处于平衡态。接下来,我们对分子动力学模拟中能量低的瞬态结构进行优化,计算吸附能,并与石墨烯比较。
对PBE、PBE-D2和vdW-DF泛函,所得到的Glu在石墨二炔表面最大吸附能分别为0.54、0.90和1.14 eV (表6)。该结果表明,PBE泛函给出的吸附能明显地小于PBE-D2或vdW-DF泛函,色散力在氨基酸吸附中占了很大一部分比例。在石墨烯表面,PBE、PBE-D2和vdW-DF泛函所得到的Glu最大吸附能分别为0.43、0.84和0.94 eV (表7)。可以看出,石墨烯对Glu的吸附明显弱于石墨二炔。对于在石墨二炔表面各种不同的位置和取向,PBE-D2泛函所得到的Glu分子与石墨二炔的吸附能分布值为0.82–0.90 eV。当Glu接近石墨二炔六元环时,吸附能最大,相应构型如图7e所示。与Gly类似的是,当Glu分子的―COOH 和―NH2基团接近石墨二炔表面时的它具有较大的吸附能。在室温下,可以迅速发生在低能构型和高能构型间的转变,因为原子动能足够越过势垒。
对His分子,PBE、PBE-D2和vdW-DF泛函所得到的在石墨二炔表面最大吸附能分别为0.73、1.22和 1.46 eV (表6)。而在石墨烯表面,PBE、PBE-D2和vdW-DF泛函所给出的His最大吸附能分别为0.45、1.00和1.14 eV (表7)。可以看出,石墨烯对His的吸附弱于石墨二炔。当His分子的杂环接近石墨二炔的六元环并且―COOH和―NH2基团靠近石墨二炔表面时,体系具有最大的吸附能,如7e所示。与Gly类似,作用力主要是―COOH和―NH2基团产生的吸引力,这自来O原子与N原子强的负电性。PBE-D2泛函给出的 His分子在石墨二炔各种吸附构型的吸附能范围为1.18–1.22 eV。
对Phe分子,PBE、PBE-D2和vdW-DF泛函所得到的在石墨二炔表面最大吸附能分别为0.77、1.27和1.53 eV (表 6)。而在石墨烯表面,PBE、PBE-D2和vdW-DF泛函所给出的Phe最大吸附能分别为0.56、1.24和1.37 eV (表7)。可以看出,石墨烯对Phe的吸附弱于石墨二炔。PBED2泛函给出的Phe分子在石墨二炔各种吸附构型的吸附能范围为1.25–1.27 eV。Phe分子吸附于石墨二炔表面的最稳定结构是 Phe的芳香环与石墨二炔的六元环平行并形成AB式堆叠,如图7e所示。这个最大吸附能比Gly,Glu及His的最大吸附能都大。这种芳香环间的相互作用应来自π–π相互作用。
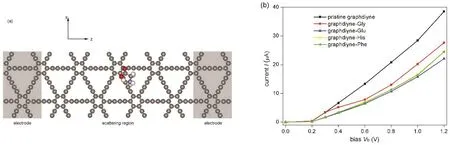
图8 (a)石墨二炔吸附Gly体系的量子电子输运计算模型,阴影区为半无限大电极,y、z方向施加周期性边界条件;(b)石墨二炔吸附氨基酸体系与纯石墨二炔的I–Vb曲线Fig. 8 (a) Calculation model of quantum electronic transport in graphdiyne-Gly system. The shadow region denotes semi-infinite electrodes. Periodic boundary conditions are applied on y and z directions; (b) I–Vb curves of graphdiyne-amino-acid systems and pristine graphdiyne.
为了考虑用石墨二炔作为生物传感器材料的可能性,我们研究了石墨二炔—氨基酸复合体系的介观电子输运性质。图8a所示为石墨二炔-Gly体系的电子输运模拟示意图,电极部分用阴影标出,在y、z方向上施加了周期性边界条件。散射区为电极原胞的 4倍大小,氨基酸分子位于散射区中间部分。在模拟中采用每种氨基酸分子最稳定的吸附构型。纯石墨二炔、石墨二炔-Gly、石墨二炔-Glu、石墨二炔-His、石墨二炔-Phe的电流电压曲线如图8b所示,都呈现出半导体特性,导通电压约为0.2 V。当电压在0.2 V以下,对于无论是否吸附氨基酸电流都趋近于零。当 Vb大于 0.2 V以后,电流随电压增大而增大。在同一Vb下,石墨二炔-氨基酸体系比纯石墨二炔的电导率明显要低。在电压Vb在1.2 V时,石墨二炔-Gly,石墨二炔-Glu和纯石墨二炔体系可由不同的电流值区分出来,然而石墨二炔—His与石墨二炔-Phe体系的电流值很接近。根据以上分析结果,不同的氨基酸可由特定电压下的不同的电流值检测出。这说明石墨二炔可被用于设计和制备生物传感器。
6 结论
石墨二炔作为一种新型二维材料,具有独特的物理性质,可用作小分子传感器的基础材料。由于石墨二炔具有一定的化学惰性,需通过掺杂来改变其分子吸附特性,提高石墨二炔对分子的响应能力。本文首先研究了石墨二炔纳米带的电子输运性质。接下来,研究了通过掺杂3d金属原子来调节石墨二炔物理性质的方法。在3d金属中,我们挑选出了在石墨二炔表面吸附能最大的Sc、Ti原子,发现掺杂Sc、Ti原子使石墨二炔能够作为气体分子检测相应器件。我们先从原子迁移势垒的角度,确定了Sc、Ti掺杂石墨二炔在室温下的稳定性。接下来,选择HCHO分子作为典型代表,从载流子浓度、介观输运等方面探讨了Sc、Ti掺杂石墨二炔对HCHO分子的响应。计算结果表明,掺杂 Sc、Ti原子的石墨二炔可高效吸附HCHO。根据掺杂改性后石墨二炔吸附HCHO分子的电流—电压曲线的改变,指出基于Sc、Ti掺杂石墨二炔可以设计二维气体分子传感器。
我们揭示氨基酸分子与石墨二炔的相互作用规律及物理机制,发现色散力在其相互作用中占主要成分。我们研究了氨基酸分子在石墨二炔表面的吸附,分析了氨基酸分子的吸附能和热运动形式。在同样电压下,石墨二炔吸附不同氨基酸后的电流值不同。该结果指明了石墨二炔作为氨基酸分子电子学检测器件的基础。本研究对今后基于石墨二炔的纳米电子学器件、生物传感器、纳米药物载体等研究提供了重要参考依据和理论指导。
猜你喜欢
杂志排行

物理化学学报的其它文章
- Theoretical Studies on the Deformation Potential, Electron-Phonon Coupling, and Carrier Transports of Layered Systems
- Advanced Progress in the Synthesis of Graphdiyne
- Chemical Modification and Functionalization of Graphdiyne
- Structure Characterization and Application of Graphdiyne in Photocatalytic and Electrocatalytic Reactions
- Graphdiyne for Electrochemical Energy Storage Devices
- Graphdiyne with Enhanced Ability for Electron Transfer