Theoretical Studies on the Deformation Potential, Electron-Phonon Coupling, and Carrier Transports of Layered Systems
2018-09-07XIJinyangNAKAMURAYumaZHAOTianqiWANGDongSHUAIZhigangMaterialsGenomeInstituteShanghaiUniversityShanghai00444China
XI Jinyang , NAKAMURA Yuma , ZHAO Tianqi , WANG Dong , SHUAI Zhigang ,* Materials Genome Institute, Shanghai University, Shanghai 00444, P. R. China.
2 MOE Key Laboratory of Organic OptoElectronics and Molecular Engineering, Department of Chemistry, Tsinghua University,Beijing 100084, P. R. China.
Abstract: The electronic structures, deformation potential, electron-phonon couplings (EPCs), and intrinsic charge transport of layered systems — the sp + sp2 hybridized carbon allotropes, graphynes (GYs) and graphdiynes(GDYs), as well as sp2 + sp3 hybridized structure with buckling, such as stanine — have been investigated theoretically. Computational studies showed that, similar to graphene, some GYs can possess Dirac cones (such as α-, β-, and 6,6,12-GYs), and that the electronic properties of GYs and GDYs can be tuned by cutting into nanoribbons with different widths and edge morphologies. Focusing on the features of Dirac cones, band structure engineering can provide a clue for tuning electronic transport in 2D carbon-based materials. Based on the Boltzmann transport equation and the deformation potential approximation (DPA), the charge carrier mobilities in GYs and GDYs were predicted to be as high as 104–105 cm2∙V−1∙s−1 at room temperature. Interestingly, due to lower EPC strength and longer relaxation time, the charge carrier mobility in 6,6,12-GY with double Dirac cones structure was found to be even larger than that of graphene at room temperature. The unique electronic properties and high mobilities of GYs and GDYs make them highly promising candidates for applications in next generation nanoelectronics. Additionally, through the full evaluation of the EPC by density functional perturbation theory (DFPT) and Wannier interpolation, the EPCs with different phonon branches and wave-vectors as well as charge carrier mobilities for graphene, GYs and stanene have been discussed. This showed that the longitudinal acoustic (LA) phonon scattering in the long wavelength limit is the main scattering mechanism for GYs and graphene, and thus the DPA is applicable. Due to stronger LA phonon scattering, the electron mobilities (~104 cm2∙V−1∙s−1) of α-GYs and γ-GYs were predicted to be one order of magnitude smaller than that of graphene at room temperature by full evaluation of the EPC. However, the DPA would fail if there was buckling in the honeycomb structure and the planar symmetry was broken (absence of σh), such as in stanene, where the inter-valley scatterings from the out-of-plane acoustic (ZA) and transverse acoustic (TA) phonons dominate the carrier transport process and limit the electron mobilities to be (2–3) × 103 cm2∙V−1∙s−1 at room temperature. In addition to our calculations, others have also found that the main scattering mechanisms in layered systems with buckling, such as silicene and germanene, are ZA and TA phonons. Thus, these results give us new insights into the role of EPCs and the limitation of the DPA for carrier transport in layered systems. They also indicate that the carrier mobilities of systems without σh-symmetry can be improved by suppressing the out-of-plane vibrations, for example by clamping by a substrate.
Key Words: Graphyne; Stanene; Electronic structure; Deformation potential; Electron-phonon coupling;Mobility
1 引言
纳米材料尤其是低维纳米材料由于其尺寸受限效应,带来了很多不同于传统三维材料的量子现象,成为电子学、材料学、光电子学等诸多领域中炙手可热的材料。在低维材料中,碳原子形成的键都比较稳定,且具有高度的灵活多样性,不同碳碳键(如 sp3、sp2和 sp杂化)形成的碳材料具有差异非常大的结构和性质,使得碳材料种类丰富多样。随着几十年来各种研究手段的不断突破和创新,人们已经能够制备出各种特点的低维碳材料,如零维富勒烯1、一维碳纳米管2、二维石墨烯3等,它们展现了优良的电子性能、电化学性能和机械性能,是能够应用于纳米电子学、化学传感器和能源存储等领域的潜在低维材料4–6。以上几类碳材料都具备sp3和sp2的杂化,然而在碳碳键中还有一种有趣的sp杂化,以sp杂化形成的碳碳三键具有线性结构、无顺反异构体和高共轭等特点,所以多年来人们也一直渴望和尝试能够获得含有sp杂化的新型碳的同素异形体。直到1987年,Baughman等7通过计算认为将炔键(C≡C)引入到苯环中形成共轭大碳环的二维平面网状结

帅志刚,1962年出生。1989年博士毕业于复旦大学。现为清华大学化学系长江特聘教授、博士生导师。主要研究领域为理论化学、材料的功能理论计算与模拟。构是可以稳定存在的,并将这类以sp和sp2杂化形成的二维碳的同素异形体称之为石墨炔,例如三种典型的含单炔键的石墨炔结构:α-,β-和 γ-石墨一炔(图1a–c);之后国际上的理论学家以及功能分子研究组都开始了相关的研究并探索制备石墨炔8–11,其中在1997年 Haley等10首次制备出了含双炔键的一种石墨炔片段,我们称之为石墨二炔(图1d);但直至2010年,石墨二炔才由中国科学院化学研究所李玉良等12利用在铜表面上通过化学原位反应的方法成功地合成了大面积薄膜,开辟了石墨炔这一重要研究领域,同时也引发了有关石墨炔的理论研究热潮。
在本文里,石墨炔是指包含了若干炔键的共轭大碳环的二维平面网状结构,碳环一边含有一个炔键的石墨炔英文名为 graphyne,我们用 GY表示(如 α-、β-和 γ-GYs),苯环间含有两个炔键的石墨炔英文名为graphdiyne,我们用GDY表示。石墨炔的合成,推动了石墨炔功能性质的研究。最新的理论研究和实验结果均显示,石墨炔在能源、环境、催化、电子等领域均显示出了许多优越性质和性能8–15。例如在电子领域,2010年 Li等12实验测试发现GDY的室温电导率为2.516 ×10−4S∙m−1,之后在 2012 年,他们制备获得的 GDY纳米线测得具有较高的室温电导率(1.9 × 103S∙m−1)和迁移率(7.1 × 102cm2∙V−1∙s−1)13。相应在理论计算方面,人们也对多种石墨炔进行了电子结构计算,并预测了它们的电荷载流子输运性质:γ-GY 和 GDY 是直接带隙半导体11,16–18,而 α-、β-和6,6,12-GYs (图1e)的能带结构具有和石墨烯类似的狄拉克锥19–21;通过形变拉伸22,23、裁剪成不同边缘形貌的一维纳米带17,24,25、加外电场26–28等手段,能够调节改变石墨炔的带边能带结构;基于玻尔兹曼输运方程和弛豫时间近似的电荷载流子迁移率计算表明这些石墨炔室温下的迁移率能 达 到 104–105cm2∙V−1∙s−116,21,25,29–31, 并 且6,6,12-GY由于存在两个狄拉克锥,使得其室温迁移率能比石墨烯还大21。

图1 常见二维石墨炔的平面结构31Fig. 1 Geometric structures of GYs and GDY 31.
由于有较多的综述文章对石墨炔进了多方面介绍14,15,32,33,在本篇综述中,我们将基于本课题组的一些工作17,21,30,31,34,从理论计算角度回顾和讨论几种典型石墨炔(α-、β-、γ-、6,6,12-GYs 和GDY)的电子结构和电荷载流子输运性质;特别地,针对迁移率计算的关键量-电声耦合(Electron-Phonon Coupling,EPC),我们将介绍高效的长声学波形变势方法(Deformation Potential Approximation,DPA)29,35和高精度的电声耦合计算方法(密度泛函微扰理论(Density Functional Perturbation Theory,DFPT)36+瓦尼尔插值(Electron-Phonon coupling using Wannier functions,EPW)37,38技术),并且通过在二维石墨烯、石墨炔和锡烯(stanene)电荷输运性质预测上的运用,表明DPA方法的适用性。
2 电子结构
2.1 二维石墨炔
从结构上讲,石墨炔分子内有三种类型的C―C键11,17,32,39:在中心芳香环上C(sp2)―C(sp2)键,键长约为 1.43 nm;连接 C=C和 C≡C的C(sp2)―C(sp)键,键长约为0.14 nm;连接C≡C的C(sp)―C(sp)键,键长约为0.123 nm,此外石墨炔具备与石墨烯相同的六边形对称结构40,41,这些几何结构特点决定了石墨炔的电子结构特征。图 2展示了目前几种常见石墨炔的基于 Perdew、Burke、Ernzerhof (PBE)泛函计算的能带结构21,42,可见:1) γ-GY和GDY是直接带隙半导体,带隙分别位于倒空间M(1/2, 0, 0)和Г(0, 0, 0)点,带隙数值分别为0.47和0.52 eV42,带隙的存在使得它们在光电器件方面有潜在的应用前景;2) α-,β-和6,6,12-GYs具有类似于石墨烯的狄拉克锥20,21:α-GY的情形与石墨烯类似,狄拉克锥点位于K(1/3, 1/3, 0)点,显示了三重对称性;β-GY的狄拉克锥点位于Γ点到M点的路径上;与六方对称性的石墨烯、α-和 β-GYs不同,两个狄拉克锥点出现在四方对称性的6,6,12-GY不可约布里渊区,其中一个狄拉克锥点位于Γ点到X’点的路径上(狄拉克锥I),而另一个狄拉克锥点位于M点到X点的路径上,并靠近高对称点X点(狄拉克锥II)。狄拉克锥I和II分别相对于Γ–X’路径和M–X路径具有镜面对称性。从图2e能明显看出,狄拉克锥I的斜率比狄拉克锥II的要大。通过对比狄拉克锥附近的斜率,发现这三类GYs的电子群速度都要比石墨烯的小,狄拉克锥的斜率与曲率半径对载流子的本征迁移率有重要的影响。

图2 基于PBE计算的石墨炔能带结构Fig. 2 Band structures of GYs and GDY at PBE level.
2.2 一维石墨炔纳米带
由于石墨烯是没有带隙的,为了将其应用于光电器件,人们通过将石墨烯裁剪成不同边缘形貌、不同宽度以及不同边缘修饰功能化的一维纳米带(NanoRibbons,NRs)来打开带隙43,44。石墨炔与石墨烯有类似的二维平面结构,人们自然想到了在石墨炔面内进行各种裁剪尝试,以此来调节能带结构17,24,25。在石墨炔中,碳碳之间存在三种成键形式,即 sp–sp、sp2–sp2以及 sp–sp2,一般认为sp–sp2之间的成键比较弱,因此,我们把sp–sp2之间的键打开,在平面内按正交的方向裁剪一定宽度的纳米带,即得到了如图 3所示的不同宽度纳米带(以 GDY 为例),上方的是扶手型纳米带(Armchair NanoRibbons,ANRs)和下方的锯齿型纳米带(Zigzag NanoRibbons,ZNRs),其中ZNRs可以有两种,即均匀带宽(如 3-ZNR)和非均匀带宽(如2.5-ZNR)的纳米带,其中数字是六边形个数,代表纳米带宽度。Du等24考察了 γ-GY和 GDY中不同宽度ANRs和ZNRs的能带结构,基于Local Density Approximation (LDA)泛函的结果表明两者的ANRs和ZNRs都是半导体,并且带隙随着纳米带宽度增加而减小,带隙数值 0.5–1.3 eV;Huang等25基于PBE泛函计算了GDY不同宽度ANRs和ZNRs的能带带隙,其数值0.48–1.54 eV。这里,我们以本课题组2011年的工作为例17,介绍GDY不同宽度ANRs和ZNRs的能带结构,并在第三部分讨论它们的电荷载流子输运性质。

图3 GDY中两种不同类型、不同宽度的纳米带(以六边形个数代表宽度),红色线框为重复单胞17 Fig. 3 Geometric structures of ANR and ZNR for
我们选取五个纳米带进行计算,如图3所示,裁剪后带边碳原子的悬挂键用氢饱和,红色线框内为重复单胞。通过PBE泛函的结构优化,ANR晶胞参数(沿带延伸方向重复单胞的宽度) a0=0.946 nm,ZNR晶胞参数为a0= 1.639 nm;2-ANR和3-ANR受限方向的宽度分别为1.25和2.07 nm,2-ZNR、2.5-ZNR和3-ZNR的宽度分别为1.92、2.39和2.86 nm。
从 PBE泛函计算的五个纳米带能带结构(见图4)中可以看出,GDY纳米带是带隙较宽的半导体。相比如二维单层GDY的0.46 eV带隙17,纳米带的带隙都达到1.0 eV左右。比较各能带的细微差别,可以发现,ANR的价带和导带带宽明显大于ZNR的能带带宽,说明GDY沿线型纳米带方向原子之间的相互作用(或称原子轨道在该方向上的成键强度)比沿锯齿型纳米带更强,这一点也不难从它们的结构图中看出:对于ANR结构,纳米带的延伸方向与两条碳原子链的排列方向一致,与另外两条成30°角;而ZNR型结构中纳米带的延伸方向与碳原子链的排列方向成 30°角或垂直。
此外,二维石墨炔及它们一维纳米带的能带结构也能通过形变拉伸22,23、加外电场26–28,45,46等进行调节,具体可以参阅相关文献。这些能带工程为石墨炔在未来纳米电子器件和光电器件上的运用打开了一条有效途径。
3 电荷载流子迁移率预测

图4 五种GDY纳米带能带结构17Fig. 4 Band structures of five nanoribbons of GDY 17.
在纳米电子器件和光电器件中,石墨烯最大的优势在于超高的电荷载流子室温迁移率达到了105cm2∙V−1∙s−13,47,这是得益于石墨烯具有狄拉克锥的电子结构。介于此,人们也在成功制备石墨炔的基础上,展开了对其电荷载流子输运性质的探索,例如:实验上在首次获得大面积二维GDY时12,测得其室温电导率 2.516 × 10−4S∙m−1;之后测得一维 GDY纳米线13具有较高的室温电导率1.9 × 103S∙m−1以及室温迁移率 7.1 × 102cm2∙V−1∙s−1;理论上,也预测表明石墨炔具有与石墨烯可比的超高迁移率17,21,29–31,具有成为下一代新型纳米电子器件的潜在材料。在这部分中,我们首先回顾计算迁移率的理论计算模型,着重介绍两种计算电声耦合的方法:长波长纵声学波形变势方法(DPA)和基于密度泛函微扰理论和瓦尼尔插值(DFPT + EPW)的精确电声耦合计算方法;然后将两种不同方法用于石墨炔电荷载流子输运性质的预测,表明石墨炔的室温迁移率可达 104cm2∙V−1∙s−1,甚至 6,6,12-GY 的迁移率能高于石墨烯;最后通过分析不同波矢、不同模式声子散射对二维平面结构石墨烯、石墨炔以及另一种二维非平面 buckling结构锡烯上的电荷载流子输运性质的影响,阐明DPA方法的适用范围,为理论工作者在计算方法选择上提供一些指导。
3.1 迁移率的理论计算模型
不同于有机材料中适用于局域电荷输运的跳跃模型和极化子模型48–50,由于碳材料中具有较高的载流子群速度、较大的能带带宽,电子态是非局域的,因此,能带模型29,49,50中的玻尔兹曼输运方程和弛豫时间近似51是计算载流子迁移率的基本手段,此时的载流子迁移率可表达为29:

其中,εnk和fnk分别表示位于第n条能带波矢为k的电子能量和平衡态下电子的费米-狄拉克分布,νnk= [(1/ћ)]▽εnk电子群速度。载流子弛豫时间τnk定义为30,52:
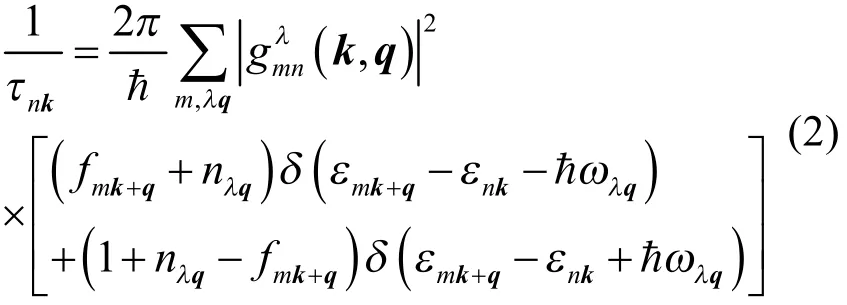
其中ћωλq和nλq分别是波矢为q、第λ模式的声子能量和对应平衡态下声子的玻色-爱因斯坦分布。第一个δ函数描述吸收声子过程,第二个δ函数描述发射声子过程。称之为电声耦合矩阵元,定义为:

这里M是原胞内原子质量,ΔλqV是声子(λ, q)振动下原子偏离平衡构型引起的晶格势场变化。电声耦合矩阵元描述了受到声子(λ, q)散射后,电子态从(n, k)到(m, k + q)的跃迁几率。
因此,计算电荷载流子弛豫时间和迁移率的关键在于求解电声耦合矩阵元。
3.2 DPA方法及其在石墨炔中的应用
第一种比较常用的方法是长声学波形变势方法(DPA),最早是在上世纪50年代由Bardeen和Shockley提出的35。DPA的出发点是由于电子在室温下的德布罗意波长约为 7 nm,远大于晶格单胞长度,因此电子主要是与描述晶格整体振动的长波长纵声学波(longitudinal acoustic,LA)相互作用。目前除了无机半导体,DPA方法在碳材料 17,21,25,29,43、有机共轭小分子29,53–55、聚合物56、有机/无机杂化钙钛矿57,58及DNA链59等体系中成功应用。DPA方法认为晶格热振动引起的晶格势场变化ΔV(r)与微小体积相对变化量Δ(r)呈线性关系,即ΔV(r)=DLAΔ(r),DLA定义为LA声子形变势常数。此时热力学平均下的电声耦合矩阵元可表示为29:


3.2.1 GDY及其纳米带
实验制备出大面积二维GDY后12,我们随即利用公式(1)和(5)计算了其室温载流子迁移率17,表明 GDY也是具有成为优秀纳米电子器件材料的潜质,其室温载流子迁移率能够达到 105cm2∙V−1∙s−1,但因其刚性较弱(弹性常数较小),迁移率比石墨烯略小;此外,基于第二部分2-ANR、3-ANR、2-ZNR、2.5-ZNR和 3-ZNR优化后晶体结构和能带结构,它们的一维弹性常数 C、有效质量m*和一维形变势常数DLA也可获得,将这些参数带入有效质量近似下一维体系迁移率计算公式29:

获得了室温载流子迁移率。表 1总结了相应的计算结果,从表中能看出:1) 无论ANRs还是ZNRs,能带带隙随着纳米带宽度增加而减小;2)ANRs电子(空穴)有效质量要比ZNRs的小;3) 两种类型纳米带的迁移率都随着带宽增加而增大,这与Huang等25的计算结果一致;4) 得益于ANRs较小的有效质量,对比宽度类似的 2-ANR与2-ZNR、3-ANR与3-ZNR,发现ANRs的迁移率要比ZNRs要大,尤其是电子迁移率;5) 由于具有较小的电子形变势常数,在所有体系中,电子迁移率要大于空穴迁移率,同时我们预测的室温一维纳米带的最高电子迁移率能达到 104cm2∙V−1∙s−1。
3.2.2 石墨炔
对于存在狄拉克锥的二维石墨炔,譬如在第二部分分析的 α-、β-和 6,6,12-GYs,由于存在较大的电子群速度,被认为能像石墨烯一样具备较强的电荷载流子输运能力,我们在2013年基于玻尔兹曼输运方程和 DPA方法,利用公式(1)和(5)预测了它们的弛豫时间和迁移率21。表 2总结了三种石墨炔以及石墨烯对应的形变势常数、二维弹性常数以及室温弛豫时间和迁移率。从表中可以看出:1) 石墨炔的弹性常数随着乙炔链的增加而减少。这是由于随着乙炔链的增长,碳原子在平面结构中变得更加稀疏,断裂应变增加,导致石墨炔硬度更低。此外,6,6,12-GY的弹性常数表现了明显的各向异性;2) 与弹性常数相似,形变势常数随着乙炔链的增加而减小。这可以归咎于sp三键比sp2双键更加牢固,因此,石墨炔在晶格形变作用下不容易发生改变;3) 随着乙炔链成分从6,6,12-GY的41.67%增加到α-GY的100%,弛豫时间也随之下降。根据公式(5)可知,在形变势常数相近时,三者弛豫时间的不同主要来自于弹性常数的差别。对于6,6,12-GY的最大弛豫时间必然产生最高的迁移率;4) 存在本征载流子迁移率比石墨烯更高的材料:6,6,12-GY,沿晶轴a方向的空穴和电子室温迁移率分别为 42.92 × 104和54.10 × 104cm2∙V−1∙s−1,大于石墨烯的 32.17 × 104和 33.89 × 104cm2∙V−1∙s−1。其原因一方面,与石墨烯相比,6,6,12-GY在a方向具有较小的弹性常数和小得更多的形变势常数,造成了6,6,12-GY的电声相互作用比石墨烯的更弱,使得6,6,12-GY沿着a方向的电子弛豫时间比石墨烯还要大;另一方面,根据公式(1),本征载流子迁移率不仅仅依赖于弛豫时间,还与群速度以及费米面的形状有关,6,6,12-GY的不可约布里渊区具有两个不等价的狄拉克锥,使其具有较高的电子群速度与电子态密度。

表1 PBE泛函计算的不同宽度ANRs和ZNRs的GDY纳米带带隙Egap、电子(空穴)的有效质量me*(mh*)、形变势常数De(Dh)、一维弹性常数C1D和室温(300 K)迁移率µe(µh) 17Table 1 Band gap Egap, effective mass me*(mh*), deformation potential constant De(Dh), 1D elastic constant C1D and mobility µe(µh) at room temperature (300 K) for five GDY NRs at PBE level 17.

表2 PBE泛函计算的α-、β-、6,6,12-GYs以及石墨烯的二维弹性常数C2D,形变势常数D,以及室温(300 K)电子(空穴)弛豫时间 τe (τh)和迁移率 µe (µh) 21Table 2 2D elastic constant C2D, deformation potential constant D, carriers scattering time τe (τh), and mobilityµe (µh) at room temperature (300 K) for α-, β-, and 6,6,12-GYs, as well as graphene at PBE level 21.
超高的迁移率使得石墨炔能够成为继石墨烯之后潜在的新型纳米电子器件材料。同时从狄拉克锥的特征出发,能带结构工程能够为二维碳材料的电子输运性质调控提供一个重要的思路。
3.3 精确电声计算及其在石墨炔中的应用
DPA方法是目前普遍处理电声耦合的有效方法,在之前关于石墨烯29,43、石墨炔21等的迁移率预测中也有效利用,但其仅仅是描述波矢 q趋于零的LA声子散射,其他波矢和模式的声子散射对二维碳材料的电荷载流子输运贡献如何?或者说DPA方法在这些材料计算中的准确性和适用性如何?为了回答这个问题,就必须精确求解电声耦合矩阵元(公式(3))。在这部分中,我们将首先介绍基于DFPT + EPW的精确计算电声耦合矩阵元的方法,之后将该方法应用于平面的石墨烯和石墨炔的电荷载流子弛豫时间和迁移率的计算,并通过与之前 DPA方法计算结果的比较,说明DPA方法的准确性和适用性30。在下一部分,我们再进一步以非平面 buckling结构的锡烯为例精确计算其电声耦合和输运性质,通过与平面石墨烯和石墨炔对比,表明DPA方法适用性与二维材料结构间的关系34。
DFPT36通过自洽求解电荷密度的一阶微扰,能够获得任意声子(λ, q)频率、本征极化矢量和对任意电子(n, k)散射的电声耦合矩阵元。但从公式(1)和(2)可见,迁移率和弛豫时间的计算需对布里渊区足够密度 k(q)进行积分,显然 DFPT的计算量是很大的,尤其对复杂体系是不能胜任的。近年来,Giustino等37,38,60基于 DFT (Density Functional Theory,密度泛函)和DFPT计算的稀疏k(q)网格中的电子能量、声子能量和电声耦合矩阵元,利用这些物理量在实空间的局域性,通过瓦尼尔插值技术获得它们在密集k(q)网格中的数值,能够在保证计算精度的同时极大提高计算效率。具体的插值过程如下:
2) 通过最大局域化瓦尼尔(Maximally Localized Wannier Functions, MLWFs)方法61,获得电子哈密顿量从倒空间到实空间的幺正矩阵{Uk},并构造实空间的电子哈密顿量:

其中 Re和 R′e是实空间中原胞坐标。实空间的声子动力学矩阵元也可通过声子极化矢量{eq}构造:

其中 Rp和 R′p是实空间中原胞坐标。实空间的电声耦合矩阵元可表示为:


通过瓦尼尔插值获得布里渊区中足够密度的电子能量,声子能量和电声耦合矩阵元后,就可以根据公式(2)和(1)分别计算载流子的弛豫时间和迁移率。
利用该方法,基于PBE泛函,我们首先预测了α-和γ-GYs的电声耦合、弛豫时间以及迁移率,同时为了对比,石墨烯也做了相应计算30。图5a–c展示了石墨烯,α-和 γ-GYs中靠近布里渊区中心的 LA声子模式对导带底电子散射的电声耦合矩阵元模的平方(石墨烯和 α-GY k = K,γ-GY k = M;m = n = 导带)。从图中可以看出,对于三种材料,布里渊区中心附近的电声耦合强度各向同性,并且两种石墨炔的LA声子作用强度都要比石墨烯强。根据形变势理论,q~0附近,声学声子波矢的模与电声矩阵元满足线性关系62:
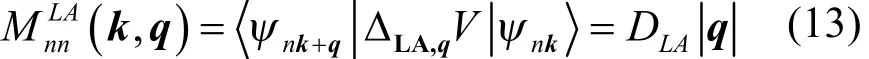
其中 DLA为 LA声子形变势常数。所以根据公式(13),可以拟合出石墨烯,α-和γ-GYs的LA形变势常数,如图5d所示。石墨烯LA形变势常数(4.24 eV)与其他第一性晶格动力学计算的结果类似(4.5 eV63、6.8 eV64),而且与通过拟合能带带边移动与形变拉伸压缩量的线性关系获得的数值(5.14 eV29)也很接近。α-和γ-GYs的LA形变势常数分别为7.34和7.77 eV,都要比石墨烯的大,表明了在石墨炔中较强的LA声子散射。

图5 石墨烯和石墨炔中LA声子散射的电声耦合强度30Fig. 5 The strength of EPC from LA phonon scatterings in graphene and GYs 30.

图6 石墨烯和GYs中的电子弛豫时间和迁移率随温度的关系30 Fig. 6 The electron scattering time and mobility as functions of temperature for graphene and GYs 30.
根据瓦尼尔插值后的布里渊区内电子能量、声子能量以及电声耦合矩阵元,利用公式(2)和(1)分别计算石墨烯,α-和 γ-GYs中载流子弛豫时间和迁移率。图6a–c展示了这三种材料中导带底的电子被平面内四种主要声子模式:纵声学波(longitudinal acoustic, LA),横声学波(transverse acoustic, TA),纵光学波(longitudinal optical,LO)和横光学波(transverse optical,TO)散射的弛豫时间与温度的关系,总的弛豫时间也通过马西森定则获得并在图中表示,图6d展示了LA声子散射下的三种材料迁移率与温度的关系;室温下四种声子模式散射下的电子(空穴)弛豫时间、迁移率以及总数值总结于表3。根据图6和表3,有以下结论:1) 常温下被LA声子散射的载流子弛豫时间(迁移率)与 DPA方法获得的结果很接近,例如精确电声计算的石墨烯和 α-GY电子弛豫时间分别为12.78和2.33 ps,电子迁移率分别为34.12 × 104和 1.07 × 104cm2∙V−1∙s−1(DPA 方法计算的数值分别为 13.22 ps29和 2.79 ps21,32.02 × 104cm2∙V−1∙s−129和 2.72 × 104cm2∙V−1∙s−121)。这说明在这类碳材料中,对LA声子散射起主要贡献是长波长(q~0)部分;2) 在很大温度范围内(包括室温)对这三种材料的载流子散射起主导作用的是 LA声子。这一方面得益于LA声子模式与电子有较强的电声相互作用,另一方面是由于在低温区和中间温区声学声子数目比光学声子要多,声学声子更易被吸收或发射;3) 在低温区,由于高频的光学声子很难被激发,其作用是可以忽略的,但随着温度升高,光学声子数目增多并由于较强的与电子间的耦合,光学声子散射作用愈发明显,尤其对于石墨烯(从图6a可见LA声子散射的曲线与LO声子散射的曲线在400 K左右发生了交叉);4) 两种石墨炔的室温迁移率都比石墨烯小了至少一个数量级,接近 104cm2∙V−1∙s−1,其原因在于对迁移率起主导作用的 LA声子散射在石墨炔中较强;5) 用DPA方法预测的石墨烯与α-GY的弛豫时间和迁移率(例如文献21中α-GY电子弛豫时间和迁移率分别为 2.79 ps 和 2.72 × 104cm2∙V−1∙s−1)与通过精确电声计算预测的数值(例如α-GY电子弛豫时间和迁移率分别2.11 ps和1.03 ×104cm2∙V−1∙s−1)比较接近,表明了 DPA 方法在这类二维平面碳材料中的适用性,但从图表中也可以看到,常温时LO声子对石墨烯输运性质也有一定贡献。

表3 室温(300 K)石墨烯、α-和γ-GYs被不同声子散射的电子(空穴)弛豫时间τ和迁移率µ 30Table 3 The carriers scattering times and mobilities with different phonon scattering mechanisms for graphene,α-, and γ-GYs at room temperature (300 K) 30.
在这部分中我们运用DFPT和EPW精确计算电声耦合并预测了石墨炔的电荷载流子输运性质,表明了对输运起主导作用的是长波长 LA声子,即DPA方法是能够很好描述的。但我们也看到,光学声子散射贡献在高温下是不能被忽略的,尤其对石墨烯。为了更加普适的说明DPA方法对二维层状材料的适用性以及各种声子模式散射与材料结构间的关系,在下一节中我们将以非平面buckling结构的锡烯精确电声耦合和输运性质计算为例,并通过与平面石墨烯和石墨炔等对比加以说明。
3.4 DPA方法的适用范围
长波长LA声子的DPA方法优势在于把复杂的电声耦合矩阵元等效为与波矢 q无关的形变势常数 DLA和弹性常数 C,将弛豫时间公式(2)简化为公式(5),提高了弛豫时间和迁移率预测的效率。同时,比较精确电声计算和DPA计算的石墨烯和石墨炔弛豫时间和迁移率(3.3节)也说明了 DPA方法对这些体系是适用的。但由于DPA描述的是q趋于零0的LA对载流子散射,忽略了声子色散以及其他声子模式对载流子散射的贡献,必然对某些体系的输运性质描述是不够的,例如:1) 无法考虑近简并能带间的载流子散射,但 DFPT精确电声计算表明双层石墨烯中近简并导带间的电子散射比较重要65;2) 无法考虑某些波矢q声子引起的谷间散射,例如单层MoS2的导带存在能量差只有约70 meV的两个能谷,当考虑了谷间散射后,电子室温迁移率会从 320 cm2∙V−1∙s−1下降到 130 cm2∙V−1∙s−166;3) 无法考虑其他声子模式散射,如在上一节表明在高温区光学声子散射对石墨烯和石墨炔输运性质的贡献不可忽略30,并且像MoS2这类极性材料,极性光学声子散射被认为是输运的主导散射模式之一67。除了以上几点,形变势常数数值上的精确性也是一个问题,以石墨烯为例,不同实验获得形变势常数通常范围很大10–50 eV68–70,而不同理论方法计算的形变势仅仅几个eV (例如4.24 eV30,4.5 eV63,5.14 eV29),数值上的差别会很大程度上影响迁移率的预测。为了更加深入说明DPA方法在二维材料中的适用性,尤其为了探索不同声子模式散射强度与材料本身结构上的关系,在这部分里,我们以非平面结构的锡烯(stanene)为例(buckling结构,即相邻原子不在一个平面上),通过DFPT和EPW精确计算不同波矢的所有声子模式对其电荷载流子输运性质的影响,并与上一节中平面结构的石墨烯和石墨烯对比,归纳总结出适合应用DPA方法的二维材料结构特点34。
锡烯是理论预测具有拓扑绝缘体特点的二维层状材料,具有独特的电子特性71–73。从几何结构上说,锡烯具有类似石墨烯的蜂窝状结构,但不同之处在于其并不是严格意义上的平面结构,相邻原子在垂直方向有0.085 nm的距离34,类似于塔扣(bucklings)的结构,如图7a所示。图7c展示了PBE泛函计算的锡烯和石墨烯的能带结构和声子结构,可见锡烯在K(1/3, 1/3, 0)点也有类似于石墨烯的狄拉克锥点,但考虑自旋轨道耦合后 K点会产生76 meV的带隙,同时看狄拉克锥点附近的E(k)~k关系,锡烯的电子群速度要比石墨烯的小;从声子谱(图 7d)上,由于锡原子质量比碳原子大,锡烯声子频率要比石墨烯小很多。
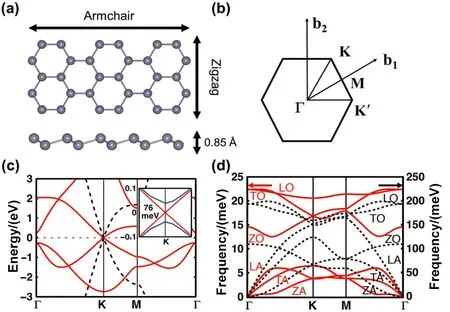
图7 锡烯几何结构、能带结构和声子谱 34 Fig. 7 Geometric structure, band structure and phonon dispersion of stanene 34.
为了说明不同模式及不同波矢 q声子散射对输运性质的影响,基于DFPT和EPW,300 K下锡烯被不同模式声子散射的谷内和谷间散射率(弛豫时间的倒数)可以根据公式(2)获得,总结于表4(作为对比,石墨烯相应也做了计算)。从表中可以看出,对于LA声子散射,无论锡烯还是石墨烯,都是以谷内散射为主,并且该散射机制决定了石墨烯的总散射率。这是由于石墨烯LA声子对应 q在布里渊区中心附近处电声耦合较强(图 8b)且声子频率相对光学声子小,对应300 K下LA声子数较多;与此不同,对比发现决定锡烯总散射率的散射机制是ZA和TA声子的谷间散射(其中ZA是垂直于平面的声子模式,out of plane acoustic),并且它们的散射率要比石墨烯LA谷内散射率几乎大一个数量级,这就会导致锡烯的迁移率比石墨烯小。其原因是由于锡烯中ZA和TA对应 q在布里渊区边界处电声耦合较强(图 8a)且它们的声子频率比石墨烯小,对应声子数更多。对比图9a锡烯和图9b石墨烯被不同模式声子散射的电子弛豫时间与温度关系也能看出,由于较强的电声耦合且较小的声子频率,使得在很大温度范围内对锡烯中载流子输运起主导作用的是ZA和TA声子散射,Li等66计算也发现对另一种buckling结构硅烯也是ZA和TA声子散射主导了总散射率;而石墨烯由于较强的电子与LA声子相互作用及LA较小声子频率,在中低温度区间的载流子输运以LA声子散射为主,高温时由于较多光学声子被激发且较强的电子与光学声子相互作用,光学声子散射起了主导。因此,仅依靠 DPA方法计算长波长 LA声子散射对锡烯输运性质的影响并不能完全反映锡烯中真实的声子散射情况。
这里需要说明的是,对谷内散射起主导作用的声学声子主要是长波长声子,由于 q~0附近的声学声子频率趋于零会导致电声耦合数值(公式(3))发散,我们默认将这些声子频率接近于零对应的电声耦合设置为零,因此会使得声学声子谷内散射被低估,尤其ZA模式。但这不影响目前的结论,即锡烯具有较强的ZA和TA谷间散射以及石墨烯以 LA谷内散射为主。我们注意到,根据Mermin-Wagner定理74,75,对于不具备 σh对称性的buckling体系(如硅烯、锗烯),具有较强的ZA声子谷内散射;Gaddemane等76表明在硅烯和锗烯中,ZA有较大的谷内和谷间散射形变势常数,而TA谷内散射形变势常数比ZA小了一个多数量级,最终是声学声子谷间散射限制了迁移率数值。这也跟我们锡烯结果类似,即TA谷内散射较弱,ZA和TA谷间散射是载流子输运的主要机制。但由于我们低估了声学声子谷内散射(尤其ZA),这需要我们今后进一步考察锡烯中ZA和TA谷内散射,并且将其与自身谷间散射、硅烯和锗烯中声子散射等比较,来说明这些 buckling体系中电声耦合与输运性质的共性与区别。

表4 室温(300 K)下锡烯和石墨烯狄拉克锥点电子被不同模式声子散射的谷内和谷间散射率34Table 4 Intervalley scattering and intravalley scattering for electrons in stanene and graphene at Dirac point K and 300 K 34.

图8 锡烯和石墨烯导带底狄拉克锥点电子与不同声子模式及不同波矢q相互作用的电声耦合强度等高图34Fig. 8 Strength of electron-phonon coupling as a function of phonon wavevector q at Dirac point K in the conduction band 34.

图 9 锡烯和石墨烯中不同声子模式散射的电子弛豫时间与温度的关系图34Fig.9 Temperature dependence of scattering time for the conduction band for all phonon modes 34.
基于目前的计算结果及相关文献报道,我们将锡烯、石墨烯以及常见二维层状材料锗烯、硅烯、α-GY和单层二硫化钼的buckling高度数值、形变势常数DLA、以及室温DPA方法计算的迁移率 µDPA(基于公式(1)和(5))和精确电声计算的迁移率 µEPC(基于公式(1)和(2))列于表 5。从表中可见buckling数值越大,形变势常数越小,对应 µDPA数值越大,同时具有buckling的体系µDPA数值比精确电声计算的µEPC数值大了好几个数量级,表明DPA方法无法全面描述这些体系输运性质,相反buckling为0的石墨烯和α-GY的µDPA和µEPC数值很接近,DPA方法是合适的。但这并不能说材料平面性越好,DPA方法的适用性越好,譬如非平面三明治结构的单层 MoS2,其 µDPA和 µEPC数值也是很接近。这是因为起主导的散射是 LA声子66,67和光学声子67,而非ZA声子散射75–77,同时在室温下LA声子数目要比光学声子数目多,所以LA声子散射主导了室温下MoS2的载流子输运。从表中声子散射模式上看,ZA和 TA在buckling结构的材料中起了主导作用,但它们并不是石墨烯、α-GY和MoS2的主要散射模式。对于ZA声子谷内散射,平面的石墨烯、α-GY和非平面三明治结构的单层 MoS2,它们都具有 σh对称性,而锡烯、锗烯和硅烯等的 buckling结构会打破这种水平镜面对称,根据Mermin-Wagner定理74,75,具有σh对称性的材料其ZA声子谷内散射是被禁阻的,相反对于不含σh对称性的buckling结构材料,由于ZA声子数目较多使得其谷内散射起了主导作用。除了ZA声子谷内散射,我们的锡烯计算结果34以及其他文献表明硅烯66,76和锗烯76都具有较强的 ZA和 TA声子谷间散射,对这种buckling结构材料的载流子输运也有较大贡献。

表5 常见二维材料buckling、形变势常数DLA、室温DPA方法计算的迁移率µDPA及精确电声计算的迁移率µEPC对比34Table 5 The buckling, deformation potential constant DLA, mobility calculated with DPA μDPA compared to that by the full evaluation of ECP μEPC for 2D materials 34.
结合表4和5的分析,我们可以得出DPA方法不适用的情况源于:1) 不具备σh对称性的二维层状体系中有较强的ZA声子谷内散射;2) 有较强的ZA和TA声子谷间散射。因此,DPA方法对平面的石墨烯、石墨炔以及非平面三明治结构的单层过渡金属硫族化合物(如MoS2) (具有σh对称性)是适用的,但对非平面具有buckling结构的硅烯、锗烯和锡烯等(不具有σh对称性)是不合适的。
4 结论与展望
在这篇综述中,我们从理论计算角度着重回顾了石墨炔的电子结构及其电荷载流子输运性质,表明有些石墨炔具有与石墨烯类似的狄拉克锥电子结构特征,例如α-, β-和6,6,12-GY;石墨炔室温迁移率能够达到 104–105cm2∙V−1∙s−1,并且由于6,6,12-GY不可约布里渊区具有两个不等价的狄拉克锥,使得在费米能级附近有较多电子分布且较高的电子群速度,加上有比石墨烯弱的LA声子散射,预测的6,6,12-GY室温迁移率能比石墨烯还高;通过将二维石墨炔沿不同方向剪切成不同宽度的一维纳米带,能够展现出不同的带隙、有效质量和迁移率数值,这为材料光电性质和输运性质调控提供了一条有效途径。在计算方法上,我们分别介绍了两种电声耦合的计算方法:DPA方法和DFPT+EPW的方法,并将它们运用到石墨炔的迁移率计算中。DPA方法的优点在于其将复杂的电声耦合等效为形变势常数和弹性常数,形式简单,是目前普遍使用的方法;后者的优点在于能够精确并高效地计算布里渊区所有波矢 q和所有声子模式的电声耦合,这为今后研究复杂体系的电声耦合及电荷载流子输运性质提供了有力的技术支持。为了说明DPA方法在二维层状材料中的适用性,我们将其与精确电声计算相比较,以平面结构石墨烯、石墨炔和非平面 buckling结构锡烯的输运性质计算为例,并结合关于非平面三明治结构MoS2和buckling结构硅烯和锗烯中电声耦合和输运性质计算的文献,表明了DPA方法在如下情况中是不适用的:1) 对于ZA声子谷内散射起主导的不具备σh对称性的二维层状材料和2)具有较强 ZA和 TA声子谷间散射的二维层状材料,例如锡烯、锗烯和硅烯等;DPA方法对具有σh对称性、以LA声子谷内散射为主的石墨烯、石墨炔和MoS2等是适用的。
这些理论预测表明低维石墨炔具备成为继石墨烯之后未来潜在的纳米电子器件材料。但同时,我们也看到目前石墨炔的研究仍处于初始阶段,大部分工作是集中在理论计算,实验上仅有GDY被成功合成,获得大面积、高质量、高稳定性的多种石墨炔材料还是存在一定困难和挑战,需要在可控制备、结构表征以及功能性质测试上继续提高;此外,新的具备结构和性能奇异性的碳的同素异形体也需要在今后结合理论和实验进行探索。我们相信石墨炔能在未来成为具有应用价值和潜力的新的碳材料,为功能材料科学的发展和进步带来新的内涵。
致谢:作者感谢龙孟秋博士对本工作早期的杰出贡献。
猜你喜欢
杂志排行

物理化学学报的其它文章
- Advanced Progress in the Synthesis of Graphdiyne
- Chemical Modification and Functionalization of Graphdiyne
- Structure Characterization and Application of Graphdiyne in Photocatalytic and Electrocatalytic Reactions
- Graphdiyne for Electrochemical Energy Storage Devices
- Graphdiyne with Enhanced Ability for Electron Transfer
- Modulation of Molecular Sensing Properties of Graphdiyne Based on 3d Impurities