分散液液微萃取-气相色谱/质谱法测定中药甘草中邻苯二甲酸酯残留
2018-09-03周艳芬贺筱雅门秀琴彭亚鸽刘万毅
周艳芬, 高 原, 贺筱雅, 门秀琴, 彭亚鸽, 刘万毅, 孟 哲
(省部共建煤炭高效利用与绿色化工国家重点实验室/化学化工学院,宁夏大学,宁夏银川 750021)
邻苯二甲酸酯类(Phthalate Esters,PAEs)化合物被广泛用于塑料制品中,以增大产品的可塑性和强度,它是一类重要的环境激素类物质。塑化剂的过多使用带来了很大的负面影响,调查表明,我国土壤PAEs的污染程度是西方发达国家的几倍到几十倍[1 - 3],而土壤中的PAEs超标会直接导致农作物及中药材中PAEs的高残留[3 - 4]。甘草是我国药食同源非常重要的一味中药材,市场需求量很大。目前市场流通的中药甘草主要来自人工栽培,但相关中药材质量控制标准还很不健全,因此加强对中药材环境污染物的监测是一项亟待解决的问题[5 - 6]。
PAEs的测定方法主要有气相色谱-质谱法[7 - 8]、高效液相色谱法[9]和液相色谱-质谱法[10]等。固相微萃取法(SPME)是近年来备受关注的新型预处理方法,但成本高,容易产生过饱和现象[11]。分散液液微萃取(DLLME)是一种采用微量体积的萃取剂,在分散剂作用下样品溶液形成乳浊现象,使目标物在样品溶液及萃取剂之间快速达到分配平衡而完成[12 - 14]。该萃取方法具有操作简单、有机萃取溶剂用量少、富集倍数高和对环境友好等优点,被广泛应用于水环境体系中农药残留及有害物质的分离富集[15 - 16]。
本文采用分散液液微萃取技术,针对不同来源的中药甘草样品,优化了提取溶剂种类、萃取剂种类和体积、分散剂体积及盐效应等前处理条件,建立了邻苯二甲酸二丁酯(DBP)、邻苯二甲酸丁苄酯(BBP)、邻苯二甲酸二环己酯(DCHP)、邻苯二甲酸二(2-乙基己)酯(DEHP)和邻苯二甲酸-正辛酯(DNOP) 5种PAEs的分散液液微萃取-气相色谱/质谱分析方法。
1 实验部分
1.1 仪器与试剂
ISQTMThermo单四极杆气相色谱/质谱联用仪(美国,Thermo Scientific),配EI源、Thermo Scientific “Xcalibur”3.1软件;QL-866旋涡混合器(海门市其林贝尔仪器制造有限公司);HGC-12A型氮吹仪(金属浴)(河北润创科技开发有限公司);250 μL的微量进样器(美国,HAMILTON公司);TGL-16M高速冷冻离心机(湘仪);HX-200型高速中药粉碎机(浙江省永康市溪岸五金药具厂)。
DBP、BBP、DCHP、DEHP、DNOP标准品(纯度大于>98%),均购自美国Sigma-Aldrich公司;甲醇、乙腈、丙酮、正己烷、环己烷(色谱纯,美国Fisher公司),二氯甲烷、氯仿、四氯化碳(色谱纯,德国MERCK公司);NaCl(分析纯,天津科密欧科技有限公司)。载气(纯度>99.9999%的氦气)购自银川市宝丰工业气体有限公司。
1.2 标准溶液的配制
用环己烷分别将DBP、BBP、DCHP、DEHP和DNOP配成浓度为100 mg/L的标准储备液;分别准确移取标准储备液,用环己烷配成浓度为10 mg/L 的DBP、BBP、DCHP、DEHP和DNOP的混合标准溶液,置于4 ℃冰箱保存,临用前以环己烷逐级稀释成系列浓度的标准工作溶液。
1.3 样品的制备
10份市售中药甘草收集自安徽亳州(AG1~AG3)、甘肃(BG1~BG3)、宁夏(CG1~CG4)。甘草样品经50 ℃干燥2 h后,粉碎至0.25 mm,袋装密封后放置在干燥器中。准确称取1.0 g甘草粉末,置于10 mL 具盖玻璃试管中,添加4 mL甲醇,密封并置于60±5 ℃水浴振荡器中1 h,涡旋1 min,3 500 r/min离心5 min。转移3.5 mL的上层清液于10 mL的玻璃离心管中,加入2.0 mL的水(分散剂)、100 μL的四氯化碳(萃取剂)和0.15 g的NaCl,振荡出现浑浊并保持1 min,3 500 r/min离心5 min;用微量注射器抽取于玻璃试管中,氮气吹干,准确加入0.5 mL的环己烷,0.22 μm滤膜过滤至进样瓶,取0.1 μL注入GC/MS仪进行测定。
1.4 气相色谱/质谱条件
色谱条件:TG -5MS毛细管柱(30 m×0.25 mm×0.25 μm);进样口温度:260 ℃;程序升温:初温为50 ℃,保持0.5 min,以30 ℃/min升至180 ℃,保持5 min,再以5 ℃/min升至290 ℃,保持5 min,最终以30 ℃/min升至320 ℃,保持5 min;载气:高纯氦气,流速1.2 mL/min;进样方式:自动,不分流进样;进样量:0.1 μL。
质谱条件:电子轰击离子源(EI),电子轰击能量70 eV;质谱传输温度280 ℃;离子源温度280 ℃;溶剂延迟时间至4.0 min;扫描模式:全扫描模式(Full)用于定性分析,其质量扫描范围m/z50~600 Da;选择离子扫描模式(SIM),通过分段选择每一个目标物的特征碎片离子进行扫描,目标分析物的保留时间、定性和定量离子见表1。

表1 5种PAEs化合物的保留时间及用于PAEs确认和测定的碎片离子

图1 不同提取溶剂对被分析物回收率的影响Fig.1 Effect of different extractants on the recovery of five PAEs
2 结果与讨论
2.1 提取溶剂的选择
实验考察了甲醇、丙酮、乙腈、甲醇∶丙酮(1∶1,V/V)和甲醇∶丙酮(1∶2,V/V)为提取溶剂时对目标物的提取效果。采用基质加标法(10 μg/L),实验结果见图1。由图1可知,甲醇对甘草中DBP、BBP、DCHP、DEHP和DNOP 5种待测物的提取回收率比其它溶剂高,其回收率均稳定在大于78.67%±5%和小于103.80%±3%之间,因此选取甲醇作为实际甘草样品中目标物的提取溶剂。
2.2 分散液液微萃取条件的优化
2.2.1萃取剂及其体积的选择取3.5 mL甲醇提取溶液,分别考察萃取剂的种类和最佳萃取体积。快速加入100 μL萃取剂和2.0 mL水及0.15 g NaCl,振荡产生浊化现象。实验结果见图2,二氯甲烷作为萃取剂,溶液不出现浑浊现象且分层效果很差;氯仿作为萃取剂,溶液出现浑浊现象,但有机相体积变化大(145±20 μL)、回收率低;四氯化碳作为萃取剂,溶液浊化现象明显,分层效果好,有机相体积约为65±5 μL,5种被测物的回收率在75.81%~103.65%之间,其相对标准偏差(RSD)小于5.61%。因此选取高密度、低水溶性的四氯化碳为样品溶液中待测物PAEs的萃取溶剂。
分别取40、60、80、100、120、140和160 μL四氯化碳考察体积用量。实验结果见图3,萃取剂体积小于40 μL,沉淀相中的四氯化碳液滴很难取出。随着四氯化碳体积的增加,样品溶液中待测物的回收率增大。当萃取剂四氯化碳的体积大于120 μL后,待测物的回收率有所下降。综合考虑四氯化碳的最佳体积选为100 μL。
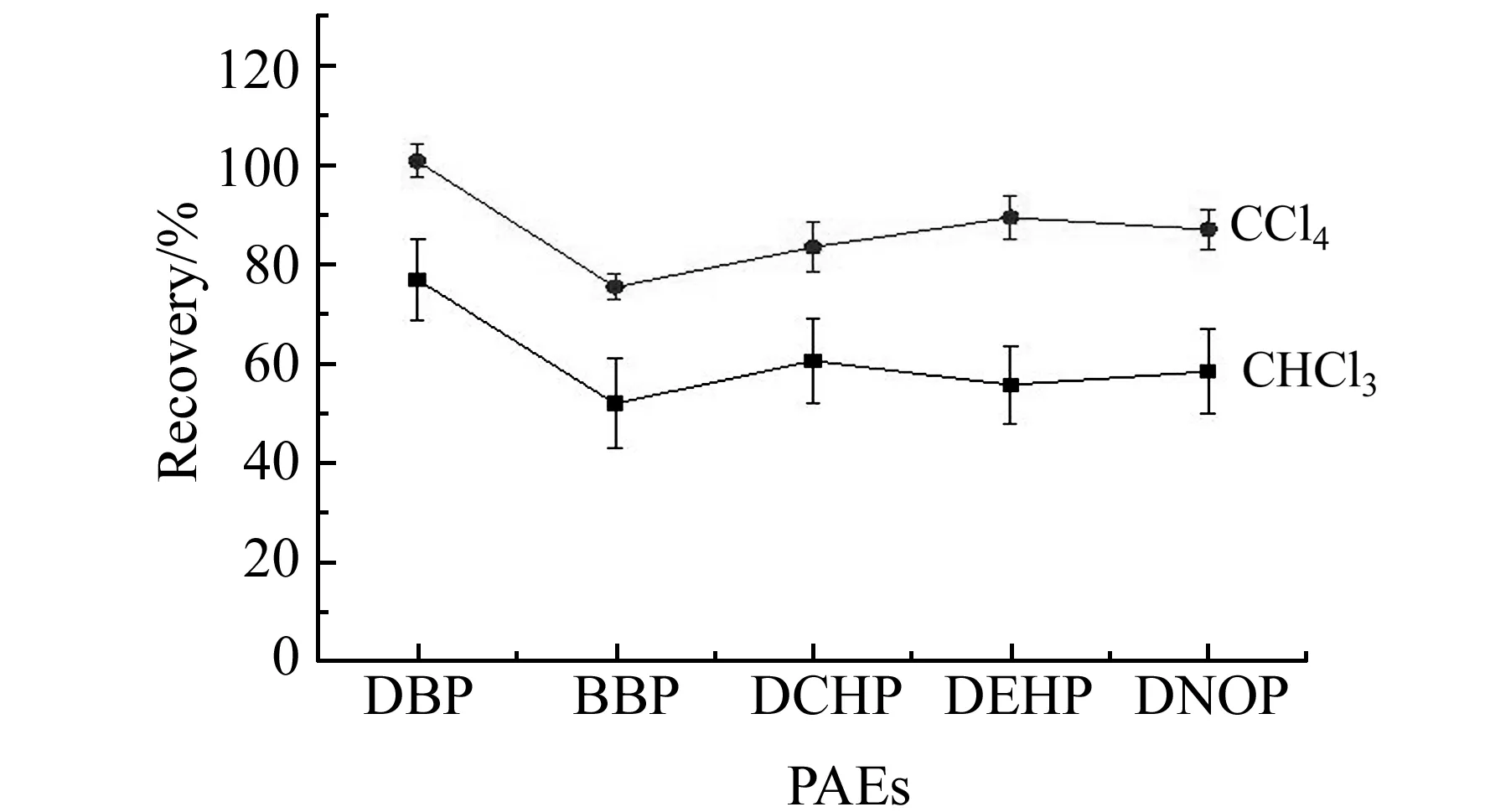
图2 不同萃取剂对被分析物回收率的影响Fig.2 Effect of different extraction solvents on the recovery of five PAEs

图3 不同体积萃取溶剂对被分析物回收率的影响Fig.3 Effect of volume of extraction solvent on the recovery of five PAEs
2.2.2分散剂及其体积的选择实验选择水为分散剂,考察了其加入体积为1.0、1.5、2.0、2.5、3.0 mL时,溶液的浊化现象及四氯化碳的提取效率。结果表明水的体积在2.0 mL溶液的浊化现象较好,DBP、BBP、DCHP、DEHP和DNOP 5种待测物的响应值较高且稳定。但随着水体积的增加,混合溶液的浊化现象消失,原因是5种待测物在体系中的分散程度增大,从而导致四氯化碳的萃取效率降低。因此,实验选取水作为分散剂,最佳体积为2.0 mL。
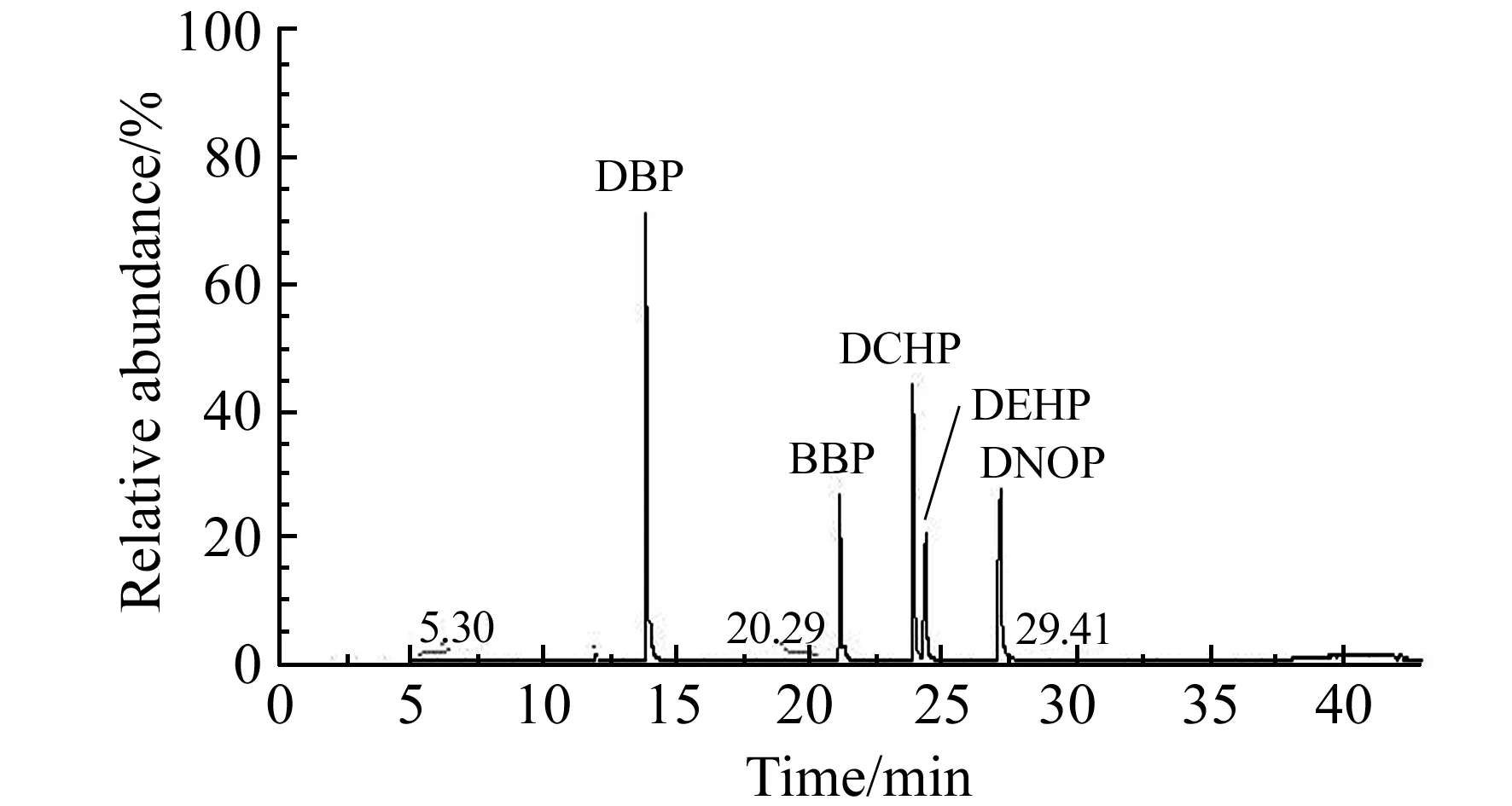
图4 5种PAEs化合物标准溶液的总离子流色谱图(TIC)Fig.4 TIC of five PAEs standard solution(concentration 10 μg/L)
2.2.3离子强度的影响溶液的离子强度主要影响目标物在分散剂和萃取剂之间的分配系数[12 - 14]。实验考察了样品中NaCl的添加量(0.05、0.10、0.15、0.20、0.25 g)对萃取效率的影响。随着NaCl加入量增加至0.15 g时,100 μL萃取剂的回收体积为65±5 μL,5种PAEs的回收率明显增加。而NaCl加入量增加至0.20 g时,100 μL萃取剂的回收体积为90±6 μL,5种PAEs的回收率略有下降。原因是随着NaCl添加量的增加溶液趋于饱和,甲醇在水中溶解度降低而使萃取剂的回收体积增加,导致待测物的浓缩倍数降低。综合考虑,选取NaCl添加量为0.15 g。在最佳的优化条件下,5种PAEs标准溶液的总离子流色谱图见图4。
2.3 线性关系与检出限
在优化分散液液萃取-GC/MS条件下,以各待测物的峰面积(y)对对应的质量浓度(x,μg/L)绘制回归曲线,结果见表2。5种PAEs在1~5 000 μg/L范围内呈良好的线性关系,相关系数均大于0.9990。该方法的检出限和定量限分别为0.16~0.58 μg/kg和0.34~1.92 μg/kg。
2.4 方法回收率与精密度
在实际的甘草样品中进行3个水平的加标回收实验。结果见表3。5种PAEs的平均回收率为87.80%~120.63%,相对标准偏差(RSD)为3.14%~7.43%(n=5)。
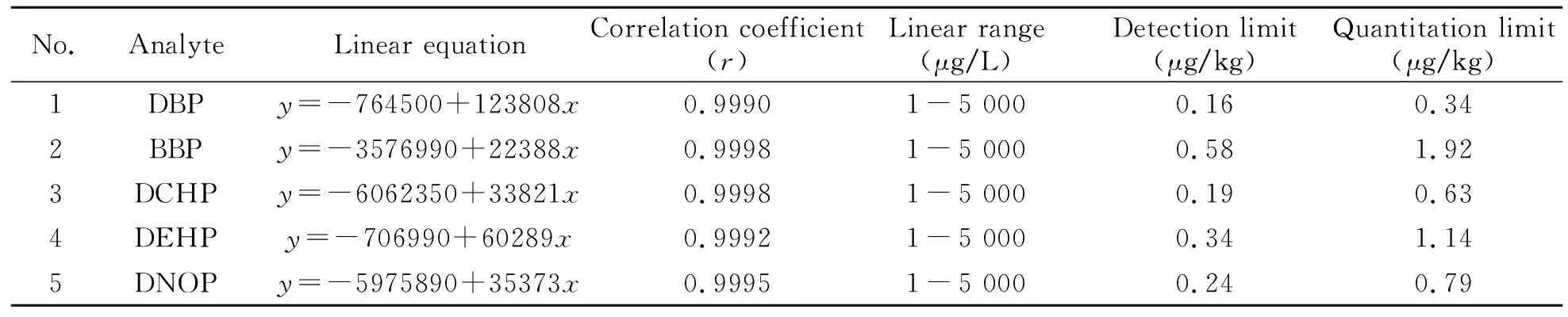
表2 5种PAEs线性关系、检出限及定量限
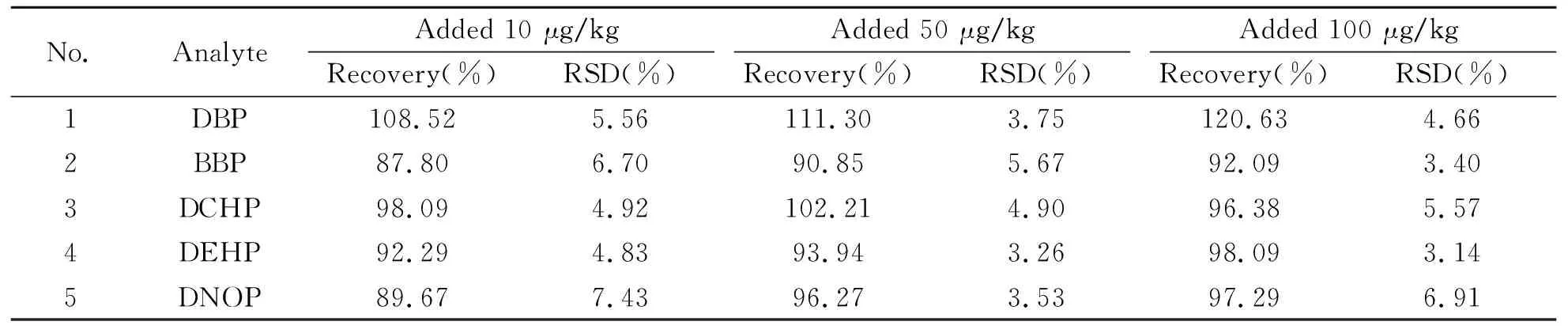
表3 5种PAEs的平均回收率及相对标准偏差(n=5)
2.5 实际样品的测定

图5 实际甘草样品中PAEs的种类和含量Fig.5 The types and contents of PAEs in licorice herbs sample(a)Blank,(b)AG1-AG3,(c)BG2-BG3 and CG1-CG3,(d)CG4 spiked.
采用本方法对三个省的10个甘草样品(AG1~AG3、BG1~BG3、CG1~CG4)进行检测,发现阳性样品需对目标物进一步确认。依据实际甘草样品CG4加标(5.0 μg/L)后如图5(d),被分析物DBP、BBP、DCHP、DEHP和DNOP的保留时间分别为13.88、21.15、23.93、24.39和27.18 min,及其相应的特征碎片离子与表1相符。结果表明所有甘草样品BG1~BG3、CG1~CG3如图5(c),AG1~AG3如图5(b),均被检测出DBP和DEHP,平均含量为0.13 mg/kg和0.09 mg/kg。三省甘草样品中5种PAEs化合物总浓度(ΣPAEs)在0.22~0.58 mg/kg范围,尤其DBP和DEHP的检出率为100%。目前,在我国甘草类中药材多为人工栽培,而对农业环境中PAEs污染特征的系统研究较少,尤其对中药材中残留的PAEs引起的健康风险缺乏关注。结合文献[1 - 3,17 - 19]的报道,土壤及设施农业环境中PAEs的污染分布与该法测定的中药甘草样品中所含PAEs的污染量成正相关性。
3 结论
建立了分散液液微萃取-气相色谱-质谱联用技术测定及确认中药甘草中PAEs组分的方法。采用该方法对10个中药甘草样品中PAEs进行测定,所有甘草药材中均被检测出DBP和DEHP,平均含量为0.13和0.09 mg/kg。三省甘草样品中5种PAEs化合物总浓度(ΣPAEs)在0.22~0.58 mg/kg范围。
