青稞种质资源遗传多样性分析与核心种质群体的构建
2018-08-28原红军曾兴权徐其君王玉林尼玛扎西
原红军,曾兴权,徐其君,王玉林,扎 桑,尼玛扎西
(1.省部共建青稞和牦牛种质资源与遗传改良国家重点实验室, 西藏拉萨 850002; 2.西藏自治区农牧科学院 农业研究所,西藏拉萨 850002; 3.西藏自治区农牧研究院,西藏拉萨 850002)
青稞(HordeumvulgareL.) 作为大麦属重要的农作物,广泛分布于全球各地,如欧洲、美国和澳洲等地,在我国主要分布在西藏及其周边地区。研究发现,西藏地区是世界栽培大麦的重要起源中心之一[1]。因其具有对高原极端气候环境的适应性,在距今约4 000年前,青稞就已作为藏区人民重要的粮食作物得到广泛种植。目前,青稞在西藏地区种植面积超过70%的耕作面积[2]。青稞的β-葡聚糖(β-glucan) 含量较高,有重要的营养价值和保健功能。
种质资源的遗传多样性和亲缘关系评估,对特异种质的选择和利用具有重要意义。在喜马拉雅地区,青稞具有丰富的种质资源,已有许多关于喜马拉雅地区的大麦资源遗传多样性的探究。在尼泊尔境内,不同青稞地方种质的农艺性状和抗病性具有显著的差异[3]。Pandey等[4]研究发现,青稞群体结构可能与资源的地理分布有关。已有研究所涉及资源群体较小,其结果可能具有一定的局限性。
尼玛扎西等[2]对西藏青稞品种拉萨勾芒进行了全基因组测序,并绘制了青稞参考基因组草图,这对青稞重要性状QTL定位、基因挖掘以及青稞遗传改良具有重要的意义。植物中常用的QTL定位方法主要是连锁分析(linkage mapping)和关联分析(association mapping),连锁分析基于双亲分离群体,关联分析则利用自然资源群体[5]。相比而言,全基因组关联分析(genome-wide association study, GWAS)的功能更为强大和高效,在较低成本条件下,可实现对QTL的高精度解析[6]。基于自然资源群体的GWAS能将鉴定到的QTL通过分子标记辅助育种,利于筛选含优良等位基因的材料,加速青稞育种进程[7]。核心种质可以较小的群体代表绝大多数遗传变异类型,是GWAS研究的重要内容之一。研究者已经利用核心自然群体对水稻[8-9]、玉米[10]、油菜[11]和花生[12]等重要农作物进行了QTL定位,发现了大量主效QTL和基因。
本研究拟利用95对SSR引物对1 220份来源广泛的青稞种质材料进行基因型分析,分析其遗传多样性及亲缘关系、群体结构及遗传分化,构建一套合适的青稞核心种质,对其更有效利用和筛选优良亲本提供参考。
1 材料与方法
1.1 试验材料
以来自全球共计22个国家和地区的1 220份青稞为材料,其中,1 055份材料来自西藏,30份来自青海,28份来自四川等地,74份国外材料分别来自欧洲、加拿大和澳大利亚等地。所有材料均由中国农业科学院国家种质资源平台提供。
1.2 试验方法
1.2.1 SSR基因型分析
对每个种质材料采用修改的CTAB(Cetyltrimethylammonium bromide)法[13],随机选择一个单株的幼嫩叶片提取DNA。利用1%的琼脂糖胶和lambda DNA,评估DNA的提取质量。利用本课题组前期开发的95对高质量SSR引物[14]对所有1 220份青稞材料进行基因型分析。PCR产物利用毛细管电泳(ABI3730 Genetic Analyzer Applied Biosystems)进行检测。采用GeneMarker V2.1软件鉴定SSR引物的不同等位变异。为了避免SSR同源片段的影响,本研究将SSR等位变异进行二进制编码(1和0),记为该等位变异出现与否[15]。每对SSR引物的统计参数为不同等位变异的平均值。
1.2.2 遗传多样性分析
统计每对SSR引物的等位变异数目(allelic count)、基因多态性(Gene diversity)、多态性信息含量(Polymorphism information index, PIC) 和香农系数(Shannon index)。其中,变异数、基因多态性和PIC值采用PowerMarker V3.51软件计算[16]。香农系数利用PopGene V1.31软件包计算[17]。考虑群体大小对多样性统计的影响,利用R程序包计算每个位点的等位变异丰度(allelic richness)。利用R程序中的t.test函数评估不同多样性参数在亚群间的差异显著性[18]。
1.2.3 群体结构和分化分析
采用STRUCTURE V2.2软件[19]对1 220份青稞材料的群体结构进行分析。假定亚群数目K为1~10,每个K值进行5次模拟运算,每次模拟进行10 000次预迭代(length of burn-in period)和10 000次马科夫链蒙特卡罗迭代(Markov chain monte carlo,MCMC),模型设定为混合和频率相关模型。依据软件输出的后验概率值[LnP(D)]和连续两个LnP(D)间的变化速率(△K)[20]确定最合适的亚群数。计算每个品种源于不同亚群的概率,当某亚群对应值最大时[21],相应品种被划分到该亚群。
利用R程序的prcomp函数对1 220份青稞材料进行主成分分析(principal component analysis, PCA),分析材料间的遗传相似性。利用R程序的Chisq.test函数来评估群体结构和材料来源(geographic origin)的相关性。采用Arlequin V3.1[22]进行分子方差分析(analysis of molecular variance, AMOVA)和pairwiseFST分析,用其评估不同亚群间的遗传分化程度,通过1 000 次排列检验来评估群体遗传分化差异显著性。
1.2.4 核心种质构建
利用R软件包corehunter[23]从1 220个青稞材料中构建一套核心种质。软件包同时考虑样本间距离(average entry-to-nearest-entry distance)和基因多态性,最优核心种质将实现有效平衡等位变异多样性和遗传差异。通过500次蒙特卡洛随机抽样,分析青稞核心种质与等样本随机群体的多样性差异。采用R程序t.test函数计算五个农艺性状(穗长、芒长、株高、穗粒数和千粒重)在核心种质和1 220份材料间的差异显著性。
2 结果与分析
2.1 群体结构和遗传分化
通过MCMC模拟,当亚群数K逐渐增加时,贝叶斯模型输出的后验概率[LnP(D)]值呈逐步增加趋势(图1),并且LnP(D)值最明显的变化发生在K由1 到2的过程中。分析贝叶斯后验概率的变化速率(△K)发现,△K在K为2时出现峰值(图 2)。综合两种方法,1 220份青稞可被划分为两个主要的亚群A1和A2(图3)。A1亚群包含192个青稞品系,其中,29个品系来自欧洲和中亚地区,15个品系来自加拿大,13个来自墨西哥,9个来自澳大利亚,其余126个品系约有117个来自西藏地区。A2亚群包含1 028个青稞品系,其中,938个来自西藏地区,52个来自青海(29)和四川(23)地区,3个来自甘肃(2)、贵州(1),约9个来自加拿大和墨西哥等地。

图1 群体结构分析中贝叶斯模型后验概率LnP(D)分布图Fig.1 Posterior probability of Bayesian model [LnP(D)] in population structure analysis
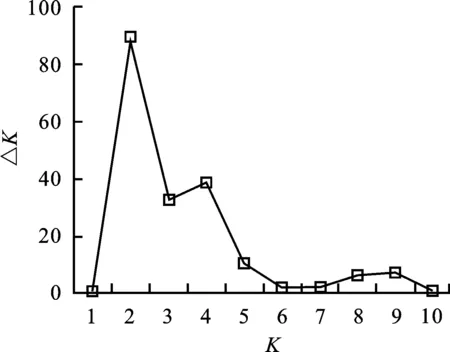
图2 贝叶斯模型后验概率变化速率(△K)分布图Fig.2 Changing rate of posterior probability(△K)
△K在K为4时出现了第二个峰值(图2),1 220份 青稞可进一步被划分为4个亚群B1~B4。B4亚群包含194个材料,包括A1亚群的192个材料以及2个加拿大材料。A2亚群则被进一步被划分为3个小亚群B1、B2、B3(图3)。通过主成分分析(PCA),第一和第二主成分(principal component)分别能解释1 220份青稞种质6.64%和4.96%的遗传变异。A1和A2亚群的青稞材料具有较明显的遗传分化,而B1~B3亚群间的遗传距离相对较近(图4)。主成分分析结果与群体材料来源具有较好的吻合度。
利用AMOVA和pairwiseFST分析青稞不同亚群间的遗传分化发现,不同亚群间遗传分化程度差异显著(P<0.001),A1和A2亚群间的遗传分化能解释17.39%的群体方差,而B1~B4亚群间的遗传分化能解释14.99%的群体方差(表1)。A1和A2亚群的分化系数FST为0.174(表2),表明我国和国外青稞种质因地理隔离和适应性等原因,具有明显的遗传差异。不同B亚群间的遗传分化程度各不相同,其中,B4与B1、B2、B3亚群的分化系数较大(FST=0.191~0.198),而B1、B2、B3亚群间的分化系数较小(FST=0.105~0.131,表2)。这表明分布于我国不同地区的青稞种质间存在一定程度的遗传差异,由于生态环境和育种需求等因素,其差异相对较小。

图3 1 220份青稞种质资源群体结构分析。Fig.3 Admixture Proportion of 1 220 hulless barley accessions
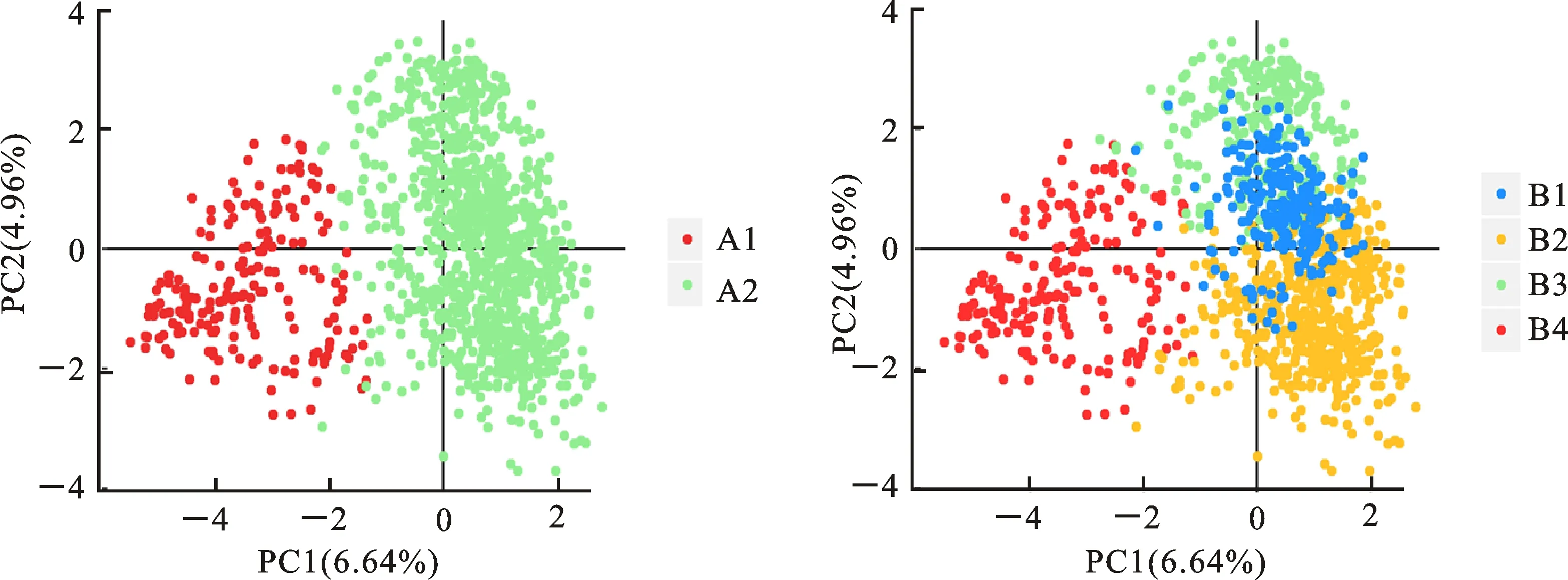
图4 青稞群体主成分分析Fig.4 Principal component analysis of hulless barley germplasm
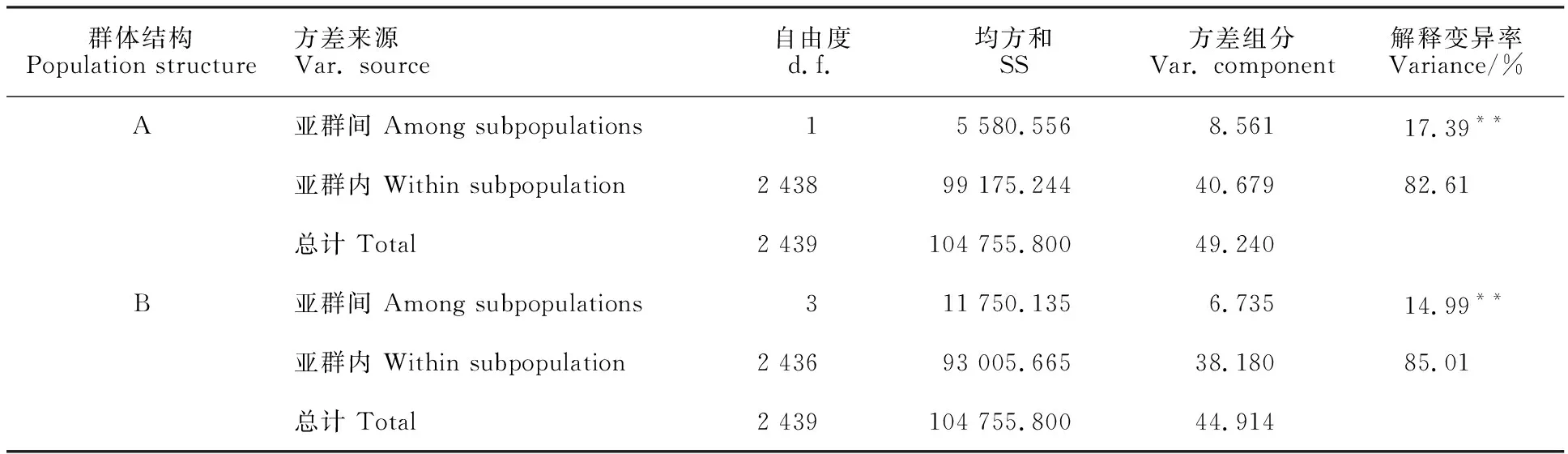
表1 青稞群体分子方差分析Table 1 Analysis of molecular variance(AMOVA) among subpopulations in hulless barley

表2 不同青稞亚群间的分化系数Table 2 Genetic differentiation(FST) between hulless barley subpopulations
2.2 遗传多样性
95对SSR引物共检测到451个等位变异,每引物平均检测到4.74个等位变异,变化范围为1~19。基因多样性平均为0.22,变化范围为0.02~0.50。多态性信息含量平均为0.18,变化范围为0.04~0.80。等位变异丰度平均为0.37(表3)。A1亚群共检测到418个等位变异,每对引物平均检测到4.40个等位基因;而A2亚群共检测到423个等位变异,每对引物平均检测到4.45个等位变异。A1和A2群体间的SSR等位变异差异不显著;其基因多样性、多态性信息含量、香农系数和最小等位基因频率均差异显著。A2亚群的群体数约为A1亚群的5倍,A1亚群的变异丰度约是A2亚群的5倍。这表明A2亚群不同材料间的等位变异冗余度较高,具有进一步筛选有代表性材料的空间。在B1~B4亚群中,B4亚群多样性显著高于其他亚群。B2亚群包括517个青稞材料,共检测到402个等位变异,等位变异丰度为0.78,约为B1和B2亚群的二分之一(表 3)。该结果表明,A2亚群中青稞材料的高冗余度主要由B2亚群材料导致。

表3 青稞不同亚群遗传多样性Table 3 Diversity statistics of subpopulations in hulless barley
2.3 核心种质构建
分析样本间遗传距离和基因多态性,从1 220个青稞材料中筛选并构建出一个青稞核心种质群体,该群体包含300份材料,其中,163个材料来自A1亚群,占A1亚群材料数的84.9%;137个材料来自A2亚群,占A2亚群材料数的13.3%。
通过蒙特卡洛模拟实验发现,从1 220个青稞材料中随机抽取300个材料能携带平均415个等位变异,变异范围为400~432,而青稞核心种质共携带444个等位变异,二者差异极显著(P<0.001)。青稞核心种质的基因型多样性指数平均为0.235,极显著高于随机群体(P<0.001)。青稞核心种质材料间的欧氏距离平均约为11.4,比原始群体间的欧式距离(8.2)明显升高(图5a)。说明该青稞核心种质能有效地代表1 220份原始材料。结合群体结构来看,在A1和B4亚群上,核心种质的遗传多样性与原始群体没有明显差别,核心种质的多样性提升主要来自于A2亚群的变化(图5b)。与此同时,最小等位变异频率在核心种质中也同样存在大幅提升,这也是群体多样性增加的一个重要原因。
青稞核心种质的穗长、芒长、株高、穗粒数和千粒重均值与原始群体并无显著差别(表4,P>0.05)。这表明青稞核心种质构建并未影响原始群体的表型分布。青稞核心种质的芒长和株高的标准差为2.89和21.49,分别为原始群体的95.7%和94.8%,而穗长、穗粒数和千粒重的标准差为1.82、18.62和6.03,为原始群体的1.08~1.18倍。该研究结果表明,青稞核心种质保留了原始群体绝大多数的表型变异,并因有效去除冗余材料,更有利于育种工作进行。

a:核心种质和原始群体的欧式距离分布图;b:核心种质和原始群体不同多样性参数的比较a:The distribution of Euclidean distances for core set and raw set;b:Comparison of genetic diversity between core set and raw set.

表4 青稞核心种质与原始群体的农艺性状比较Table 4 Comparison of agronomic traits between hulless barley core set and raw population
3 讨 论
国内外有众多研究者对不同来源的青稞种质的遗传多样性进行过研究。杨 菁等[24]利用7个SSR引物对青海省42份栽培青稞进行遗传了多样性分析,每个SSR引物平均检测到3个等位变异。范志芬等[25]利用30个SSR引物对64份青藏高原栽培青稞种质进行分析,发现栽培青稞有丰富的遗传多样性,平均每个SSR引物检测到4.4个等位变异。杨 平等[26]对四川高原的25份青稞育成品种进行SRAP分子标记分析,每个SRAP平均检测到5.2个等位变异,表明四川高原的青稞具有较高的遗传多样性。Pandey等[4]利用42个SSR引物对107个尼泊尔青稞材料进行基因型分析,发现尼泊尔青稞种质也具有较高的遗传多样性。
本研究中,1 220份青稞材料共检测到451个等位变异,每个SSR引物平均检测到4.7个等位变异。该结果比SRAP多态性水平略低,这可能是不同分子标记的扩增效率差异导致的。不同研究差异的可能原因如下:1)不同研究所用SSR引物的差异;2)扩增与检测手段存在差异;3)研究群体遗传背景的差异。随着青稞参考基因组的公布和高通量基因型分析技术的发展,大麦已经开发出基于Illumina平台的9K iSelect SNP芯片。这预示着高通量SNP分析技术的发展,对青稞的基因型鉴定、遗传多样性分析和重要性状关联分析具有重要的意义。
在农作物育种中,核心种质的筛选对杂交亲本组配和大规模种子库保种具有重要意义。核心种质能在一个较小的群体中最大化保留该群体的遗传多样性,从而提高育种效率。本研究从广泛收集的1 220份青稞种质资源中,通过评估其遗传多样性和群体结构,构建了一个青稞核心种质群体。该青稞核心种质群体保留了原始群体98%的遗传变异信息,极大地降低了材料间的冗余度,显著提高了群体多样性水平。除此之外,本青稞核心种质显著地提升了群体最小等位基因频率(MAF),这为后续基于核心种质的GWAS研究以及分子育种奠定了坚实的基础。
