Genetically confirmed familial hypercholesterolemia in outpatients with hypercholesterolemia
2018-08-17XuWANGLongJIANGLiYuanSUNYueWUWenHuiWENXiFuWANGWeiLIUYuJieZHOULuYaWANG
Xu WANG, Long JIANG, Li-Yuan SUN, Yue WU, Wen-Hui WEN, Xi-Fu WANG, Wei LIU,Yu-Jie ZHOU, Lu-Ya WANG
Genetically confirmed familial hypercholesterolemia in outpatients with hypercholesterolemia
Xu WANG1, Long JIANG2, Li-Yuan SUN3, Yue WU2, Wen-Hui WEN2, Xi-Fu WANG1, Wei LIU4,Yu-Jie ZHOU4, Lu-Ya WANG2
1Emergency & Critical Care Center, Beijing Anzhen Hospital, Capital Medical University, Beijing, China2Beijing Anzhen Hospital, Capital Medical University, Beijing Institute of Heart Lung and Blood Vessel Disease, the Key Laboratory of Remodeling-related Cardiovascular Disease, Ministry of Education, Beijing, China3Department of Dermatology, Beijing Anzhen Hospital, Capital Medical University, Beijing, China4Department of Cardiology, Beijing Anzhen Hospital, Capital Medical University, Beijing, China
Familial hypercholesterolemia (FH) is an autosomal dominant disorder of lipoprotein metabolism which can lead to premature coronary heart disease (pCHD). There are about 3.8 million potential FH patients in China, whereas the clinical and genetic data of FH are limited.Dutch Lipid Clinic Network (DLCN) criteria was used to diagnose FH in outpatients with hypercholesterolemia. Resequencing chip analysis combined with Sanger sequencing validation were used to identify mutations in the definite FH patients according to DLCN criteria. In silico analysis was conducted in mutations with previously unknown pathogenicity. Then, the novel mutant receptors were transfected into human embryo kidney 293 (HEK-293) cells. The binding and the internalization activities of the mutant receptors were analyzed by flow cytometry.The prevalence of definite FH in outpatients with hypercholesterolemia in this study is 3.2%. Using genetic testing, one homozygous FH (HoFH), one heterozygous FH (HeFH) and three compound heterozygous FH patients were confirmed. Eight mutations in low-density lipoprotein receptor (LDLR) gene were identified, in which c.357delG was a novel mutation and co-segregated with the FH phenotype. Bioinformatic analysis confirmed that c.357delG was a pathogenic mutation. Furthermore, when compared with the wild-type LDLRs by flow cytometry analysis, the binding and internalization activities of c.357delG mutant LDLRs were reduced by 35% and 49%, respectively.This study identified eight LDLR gene mutations in five patients with definite FH, in which c.357delG is a novel pathogenic mutation. These findings increase our understanding of the genetic spectrum of FH in China.
J Geriatr Cardiol 2018; 15: 434440. doi:10.11909/j.issn.1671-5411.2018.06.006
Familial hypercholesterolemia; Low-density lipoprotein receptor; Mutation
1 Introduction
Familial hypercholesterolemia (FH; MIM: #143890) is an autosomal dominant genetic disease characterized by high levels of low-density lipoprotein cholesterol (LDL-C), cutaneous xanthomas and premature coronary heart disease (pCHD), which has recently become a big concern in the world.[1]Four genes have been identified as being responsible for FH, namely, LDL receptor (LDLR), apolipoprotein B (ApoB), proprotein convertase subtilisin/kexin-9 (PCSK9) and LDLRAP1. Mutations in LDLR are the most common cause of FH followed by mutations in ApoB and PCSK9.[2]
The LDLR gene lies on the short arm of chromosome 19 at 19p13.1-p13.3, spans 45 kb and consists of 18 exons and 17 introns.[3]The LDLR gene encodes the LDLR protein, which contains 843 amino acids.[4]So far, over 1700 mutations in the LDLR gene have been described in FH (www.ucl.ac.uk/ldlr/LOVDv.1.1.0/ and https://grenada.lumc. nl/LOVD2/UCLHeart/home.php? select_db = LDLR),including large fragment deletions, small fragment deletions, insertions, point mutations and splicing mutations. Different kinds of mutations in the LDLR gene generate malfunction of the protein. As a result, clearance of LDL-rich particles from the circulation decreases, and the elevated blood serum LDL-C levels cause early onset of atherosclerosis and an increased risk of pCHD in patients with FH.[5]
It is largely believed that in the general population, the prevalence of homozygous FH is 1/1,000,000, while the prevalence of heterozygous FH is 1/500. However, the heterozygous FH frequency is even higher than 1/500 in some populations, and the elevated frequency is generally due to a founder effect.[6]FH is under-diagnosed and under-treated in the general population worldwide and no more than 1% FH patients are diagnosed in most countries.[7]Hitherto, only one study has estimated the prevalence and treatment of FH in China, which included 9324 subjects from the Jiangsu Nutrition Study. The researchers reported that the prevalence of probable/definite FH was 0.28% (1.4/500) based on the modified Dutch Lipid Clinic Network (DLCN) definition. Only 15.9% of the patients received drug treatment, and none of them achieved the LDL target value. This study suggested that FH is also under-diagnosed and under- treated in China.[8]
With the advances in genetic testing, direct detection of mutations in candidate genes is now available. In this study, we used DNA re-sequencing array technique combined with Sanger sequencing validation to identify mutations in clinical FH patients. We also evaluate the pathogenicity of the mutations found in this study.
2 Methods
2.1 Study subjects
We studied 156 hypercholesterolemia patients attending the lipid clinic of Anzhen Hospital in Beijing, China over two 6-month periods from April 2014 to June 2016. Patients diagnosed as definite FH were enrolled for genetic testing according to the Dutch Lipid Clinic Network (DLCN) Criteria.[7,9]Secondary hypercholesterolemia, such as hypothyroidism, renal or hepatic disease, were excluded by laboratory tests. DNA samples from patients with definite FH were processed using a DNA sequencing array and validation by Sanger sequencing. Some pro-bands were reported in previous studies.[10]
The study was reviewed and approved by the Ethics Committee of the Beijing Anzhen Hospital. Written informed consent was obtained from all research subjects and their legal guardians.
2.2 Measurement of serum lipids
Peripheral venous blood samples were drawn from the pro-bands and available family members after a 12-h fast. Plasma total cholesterol (TC), LDL-C, triglyceride (TG) and high-density lipoprotein cholesterol (HDL-C) were measured using routine commercial kits (Beckman Coulter, Brea, USA) and an automated biochemistry analyzer (Beckman AU 4500, Brea, USA).
2.3 Identification of LDLR mutations
A re-sequencing microarray chip was used to identify candidate gene mutations from DNA samples provided by the five probands. The resequencing array was designed on the basis of photolithography and solid-phase DNA synthesis by Vita Genomics, Inc. and manufactured by Affymetrix (Santa Clara, CA).[11]Each micro-array contained 12.6 kb of coding exon and flanking intron sequences (both sense and antisense) of the three most relevant FH causing genes, LDLR, ApoB, and PCSK9, in duplication.[11]The FH arrays were scanned using the Affymetrix GeneChip scanner 3000 7G creating CEL files for subsequent analysis. According to the results of the microarray analysis, we amplified several mutated DNA fragments that would result in amino acid changes. Sanger sequencing was conducted to verify these mutations. Family verification was conducted by sequencing the DNA of both parents at the target segment. Novel mutations were tested in 50 unrelated healthy individuals. To confirm the heterozygous deletion mutation, clone sequencing analysis was conducted. T4 DNA ligase was used to ligate the target fragment to T vecto. Then the target fragment was transformed into Escherichia coli. Further the positive clonal were picked up and cultured. Plasmid extraction was conducted, and finally the desired fragment was amplified by PCR and sequenced.
2.4 Prediction of mutation effects
Novel mutations found in this study were evaluated by searching the PubMed and LOVD databases (www.ucl. ac.uk/ldlr/LOVDv.1.1.0/ and https://grenada.lumc.nl/LOVD2/ UCLHeart/home.php?select_db = LDLR). Pathogenicity prediction of the new mutations found in this study was assessed in silico using the following publicly available software: PolyPhen2,[12]SIFT,[13]and Mutation Taster.[14]The reference sequence used for LDLR was NM_ 000527.3 (NCBI RefSeq). The online SWISS-MODEL was taken to predict structural changes of mutant proteins (http://swissmodel.expasy.org/).
2.5 Construction of eukaryotic expression vectors for LDLR
Full-length human wild-type LDLR cDNA was inserted into pReceiver-M29 to construct an LDLR recombinant eukaryotic expression vector with an enhanced green fluorescent protein (eGFP) tag. DNA encoding the c.357delG mutant of the LDLR was made using the QuickChange XL system (Stratagene, USA) and verified by DNA sequencing. The primer sequences used to generate the 357delG mutant were: forward, 5¢-TCGCTGCCACGATGGAAGTGCATC TCTCGG-3¢; reverse, 5¢-CCGAGAGATGCACTTCCAT CGTGGCAGCGA-3¢. cDNA for the wild-type LDLR and c.357delG and W462X LDLR mutants was transfected into HEK-293 cells using Lipofectamine-2000 (Invitrogen, USA).
2.6 Flow cytometric analysis
Flow cytometry was used to detect the amount of cell surface LDLR binding and LDL internalization. The fluorescence of 10,000 events for each sample was acquired for data analysis. Forward scatter versus side scatter gates were set to exclude dead cells and debris. Experiments were repeated at least three times with triplicate samples for each cell line.
To measure LDLR binding and internalization activity, the cells were transfected with wild-type, W462X and c.357delG LDLR were incubated in serum-free medium containing 20 mg/mL Dil-LDL for 4 h at 37°C. After the LDL incubation, the medium was removed, and the cells were cultured to perform the functional studies. The amount of LDL binding and internalization was detected using a FACS Calibur cell analyzer (Beckman Coulter).
3 Results
3.1 Clinical features
According to the Dutch Lipid Clinic Network Criteria, five patients were diagnosed as definite FH. The prevalence of definite FH in patients with hypercholesterolemia in this study is 3.2%. Clinical and biochemical characteristics of the pro-bands are shown in Table 1. All the pro-bands pre-sented xanthoma and two of them had corneal arcus. TC and LDL-C were extremely elevated and TG was normal before treatment. Color Doppler ultrasounds showed that every pro-band had varying degrees of mitral or aortic valve regurgitation.
3.2 Identification of gene mutations
DNA samples were analyzed by a DNA resequencing array (Figure 1). One HoFH, one HeFH and three compound heterozygous FH patients were confirmed. Overall, eight different LDLR mutations were found in the five probands (Table S1). No mutations in ApoB and PCSK9 genes were found in any of the probands. Among the eight mutations detected, there were five missense mutations, two nonsense mutations, and one frameshift mutation.
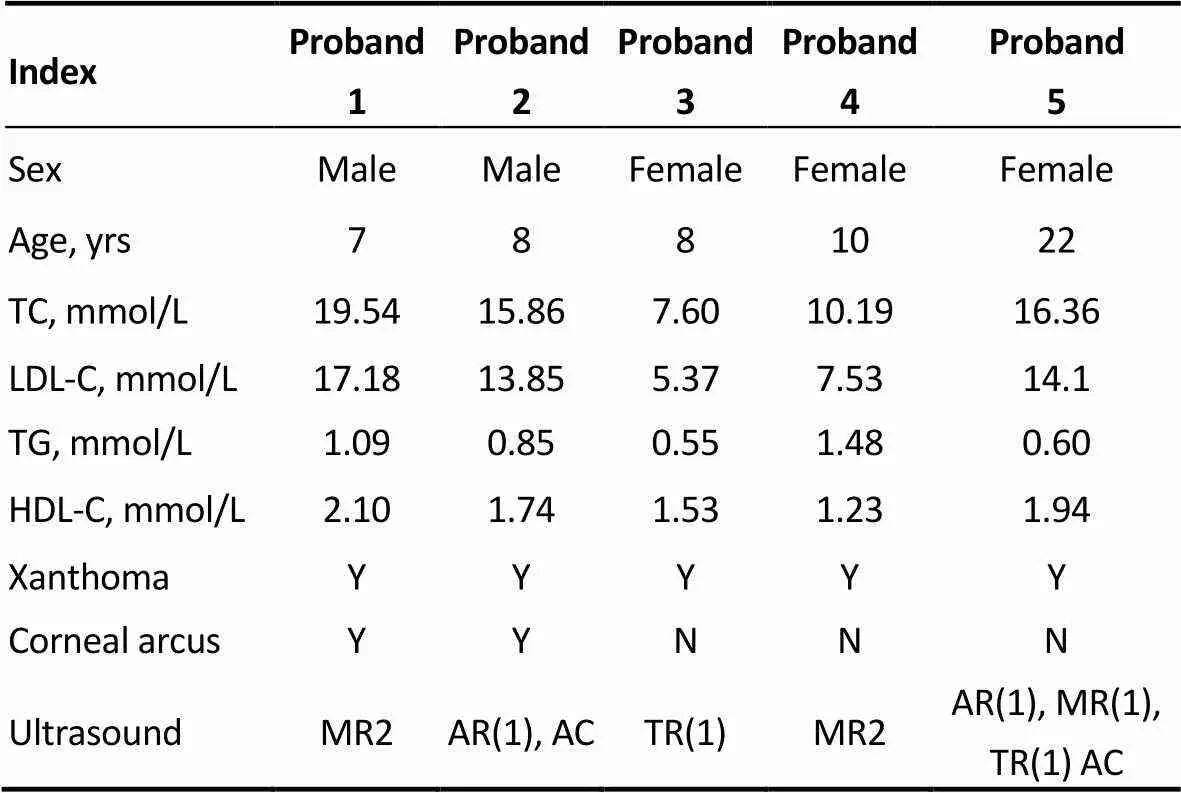
Table 1. The clinical features of five definite FH probands.
ACaortic calcification; ARaortic regurgitation; FH: familial hypercholesterolemia; HDL-Chigh-density lipoprotein; LDL-Clow-density lipoprotein cholesterol; MR: mitral valve regurgitation; N: no; TCtotal cholesterol; TGtriglyceride; TRtricuspid valve regurgitation; Y: yes.
A novel heterozygous deletion mutation (c.357delG) in exon 4 of the LDLR gene was found in proband III. Consistent with the data from the micro-array analysis, Sanger sequencing analysis of proband III revealed a heterozygous single nucleotide deletion (c.357delG) in exon 4 (Figure 2). Clone sequencing analysis verification the single heterozygous nucleotide G deletion. The heterozygous mutation was also present in another six members of her family who had hypercholesterolemia (Figure 3), and healthy members do not carry the mutation. Therefore, this mutation co-segregated with the FH phenotype. We also genotyped 50 unrelated healthy individuals as controls and did not find the mutant allele in any of these individuals.
3.3 Pathogenicity mutations found in the LDLR gene
The new mutation (c.357delG), which is absent from published controls, confers a frameshift mutation Gly119fs *86 with the new reading frame ending in a stop codon at 86 amino acid sites after the deletion site. Thus, this frameshift will cause a genetic loss of function. According to the three software tools, in silico analysis predicted this mutation to be pathogenic (Table S2). The protein structure prediction of c.357delG mutation by Swiss model showed no protein structure occurred at 86 amino acid sites after the deletion site (Figure S1).
S565X(c.1757C>A, p.Ser586X)[15], W462X(c.1448G>A, p.Trp483X)[16]and Q12X(c.97C>T, p. Gln33X)[5,16-20]also introduce premature stop codons leading to truncation of the LDLR protein which can severely damage the expression and function of LDLR. A606T (c.1879G>A, p.Ala627Thr),[16]L561F(c.1744C>T, p.Leu582Phe),[10]C122Y(c.428G>, p.Cys143Tyr)[21]and C152R (c.517T>C, p.Cys173Arg)[22]are missense variants, which have been previously reported and can lead to FH. S565X and L561F are pathogenic mutations first reported by our group that cause significantly diminished expression, binding and uptake of LDLR. A606T is one of the most common mutations among Chinese FH probands.[23]A606T resides in the thirteenth exon, which encodes the epidermal growth factor (EGF) precursor homology domain. This mutation can reduce the mobility of the mature LDLR protein, the synthesis of the precursor protein, binding of LDL, and the rate of recirculation.[16]
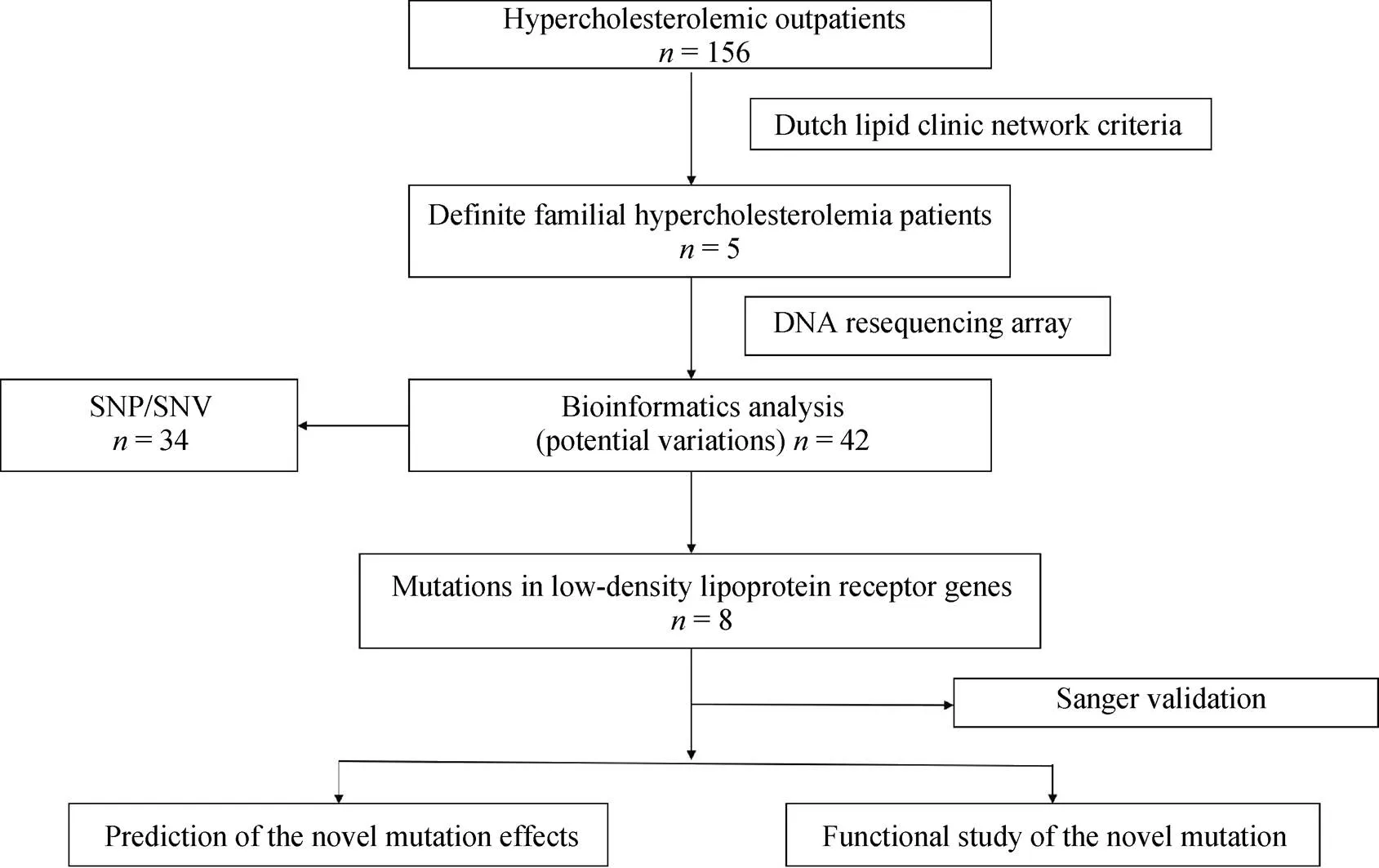
Figure 1. The flow chart of mutations screen. SNP: single nucleotide polymorphism; SNV: single nucleotide variants.
Figure 2. The DNA sequencing results of the proband 3 and his mother. (A): The LDLR gene sequencing chromatogram on exon 4 of the proband 3, the heterozygous nucleotide G deletion (shown by the arrow) leads to a frame-shift in one allele; (B): proband 3’s mother carries the same hereozgous nucleotide G deletion mutation; (C&D): clone sequencing analysis verifies the single heterozygous nucleotide G deletion. Graph D shows a DNA strand with the single nucleotide G deletion and graph C shows the normal complementary strand. LDLR: low-density lipoprotein receptor.
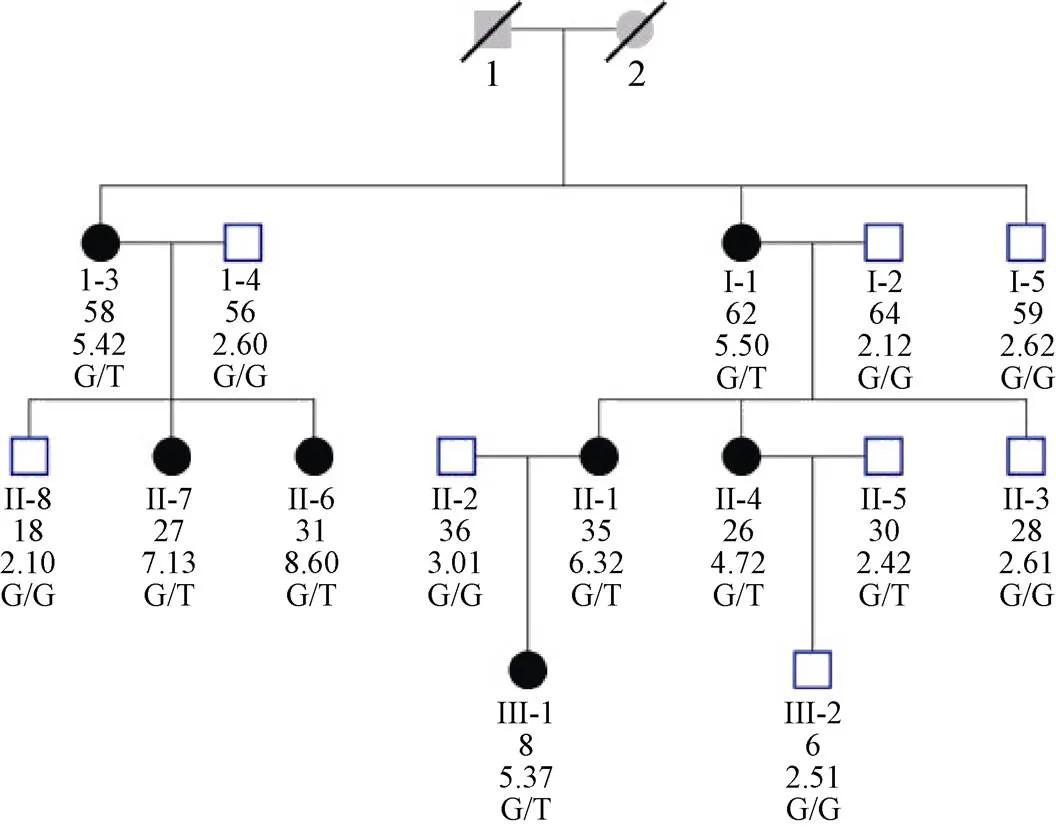
Figure 3. Pedigree chart of proband 3. Squares indicate male family members, and circles, female family members. Slashes indicate deceased persons. The column of five values under each symbol indicates, from top to bottom, the age in years (as of 2006), the TC levels (in mmol/L) and the LDLR genotypes. LDLR: low-density lipoprotein receptor; TC: total cholesterol.
3.4 The c.357delG mutant of the LDLR shows defects in LDL binding and internalization
Flow cytometry was used to observe cell-surface LDLR binding and internalization in HEK-293 cells that were transfected with wild-type or mutant LDLR (c.357delG and W462X). In comparison with the wild-type control, both the W462X and c.357delG mutants showed lower LDL binding and reduced ability to internalize (Figure 4). The binding and internalization activities of the W462X mutant, which is a known mutation, were significantly reduced to 17% binding and 39% internalizing ability. The c.357delG mutant also showed lower binding and internalization activities, reduced to 65% and 51%, of the control values, respectively (Figure 3).
4 Discussion
FH is not rare in the Chinese population and remains unidentified and under-treatment.[8]Many FH patients are not diagnosed until their first coronary events occurred.[24]Early diagnosis is critical for FH patients, because Cholesterol-lowering medication such as Statin treatment can reduced the risk of premature atherosclerosis and improved the prognosis for FH.[25]The development of next-generation sequencing technology makes rapid molecular diagnosis possible and provides a basis for personalized treatment.
In this study, five patients were diagnosed as definite FH by Dutch Lipid Clinic Network Criteria. DNA resequencing array was conducted in these patients and eight mutations include one novel mutation were identified. To ensure that this new mutation was associated with FH, verification of the c.357delG mutation was carried out by mutation allele- specific amplification in all 14 members of the proband’s family. We found that c.357delG segregated with the FH phenotype in affected family members. Furthermore, the mutation was not observed in 50 random healthy human DNA samples, indicating that it is not a common polymorphism in Chinese population. This deletion mutation (c.357delG) causes a premature stop, resulting in a truncated protein and reduce the function of LDLR. Using the three software tools, in silico analysis predicted this mutation to be pathogenic. Functional characterization of this variant was also found to be pathogenic.
We identified eight mutations in the LDLR gene of five FH patients in this study. Mutations in LDLR can occur in the promoter, introns and exons. LDLR mutations of Chinese FH patients occur mostly in the fourth exon, accounting for 19.5% of all the LDLR mutations.[16]It is generally known that high frequency LDLR gene mutations can be useful for FH screening. For instance, the S472RfsX44 mutation accounts for approximately 42% of the FH patients in Tunisia.[26]Previous studies have suggested that there is no definite common mutation in Chinese populations due to a founder effect. However, a systematic review of Chinese FH patients found that the three most common mutations: A606T, W462X and D601Y mutations are responsible for 29.3% of probands in mainland China.[23]We also identified W462X and A606T mutations in the subjects of our study.
In this study, the c.357delG LDLR mutants displayed reduced binding and internalization activities compared with the wild-type LDLR. The LDLR is a highly conserved integral membrane glycoprotein consisting of five domains: a ligand-binding domain (approximately 292 amino acids, exons 2 to 6), an epidermal growth factor (EGF) precursor homologous domain (approximately 400 amino acids, exons 7 to 14), a carbohydrate-rich domain (approximately 58 amino acids, exon 15), a transmembrane domain (approximately 22 amino acids, exon 16) and a cytoplasmic tail (ap- proximately 50 amino acids).[6]The c.357delG frameshift mutation is located in exon 4 of the LDLR gene within the precursor domain that encodes the ligand-binding domain. The mutation results in a frameshift with an expected premature stop codon and produces a truncated protein, which leads to the absence of all or part of the ligand-binding domain, including the EGF precursor homologous domain, the O-linked sugar domain, the membrane-spanning domain and the cytoplasmic tail of the LDLR. Therefore, this mutation can severely damage the function of LDLR. Two other mutations near this locus in the LDLR gene, 355del7 and 353delA, have been reported in Japanese and British patients with FH.[27,28]These mutations create premature stop codons 85 and 95 bases downstream of the deletion sites, respectively.

Figure 4. Analysis of variant functionality by FACS. The cells were incubated with PE-conjugated mouse monoclonal anti-human LDLR antibody at room temperature for 30 min. The upper right area of the dot plots represents eGFP and DiI-LDL double positive cells. (A-C): LDL binding of WT, c.357delG and W462X after 4 h incubation at 4°C; (D-F) LDL internalization efficiency of WT, c.357delG and W462X after 4 h incubation at 37°C; (G) Functional characterization of LDLR variants. 10,000 cells were acquired in a Facscalibur and values of LDL uptake were calculated as described in Materials and Methods. The values represent the mean of triplicate determinations (n ¼ 3); error bars represent SD. *< 0.05 compared to WT using a Student’s-test. eGFP: enhanced green fluorescent protein; LDLR: low-density lipoprotein receptor; PE: phycoerythrin.
In conclusion, we identified eight mutations in the LDLR gene of five FH patients, in which c.357delG is a novel mutation. The novel mutation c.357delG was found to be co-segregated with the FH phenotype. Bioinformatic analysis and flow cytometry confirm c.357delG to be a pathogenic mutation. This study increases our understanding of the mutational spectrum of FH in the Chinese population.
Acknowledgements
We greatly appreciate the support of the patients and their families for participation in this study and cardiologists who participated in this study. This work was supported by grants from the National Natural Science Foundation of China (No. 30470722, 30771982 and 30772356) and the Beijing Natural Science Foundation (No. 7032012, 7052021 and No. 7062010). The authors have no conflicts of interest to declare.
1 Watts GF, Gidding S, Wierzbicki AS,. Integrated guidance on the care of familial hypercholesterolaemia from the International FH Foundation.2014; 171: 309–325.
2 Rader DJ. Monogenic hypercholesterolemia: New insights in pathogenesis and treatment.2003; 111: 1795–1803.
3 Brown MS, Goldstein JL. A receptor-mediated pathway for cholesterol homeostasis.1986; 232: 34–47.
4 Marais AD. Familial hypercholesterolaemia.2004; 25: 49–68.
5 Gent J, Braakman I. Low-density lipoprotein receptor structure and folding.2004; 61: 2461–2470.
6 Austin MA, Hutter CM, Zimmern RL, Humphries SE. Genetic causes of monogenic heterozygous familial hypercholesterolemia: A HuGE prevalence review.2004; 160: 407–420.
7 Nordestgaard BG, Chapman MJ, Humphries SE,. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: Guidance for clinicians to prevent coronary heart disease: Consensus statement of the European Atherosclerosis Society.2013; 34: 3478–3490.
8 Shi Z, Yuan B, Zhao D,. Familial hypercholesterolemia in China: Prevalence and evidence of underdetection and undertreatment in a community population.2014; 174: 834–836.
9 Haase A, Goldberg AC. Identification of people with heterozygous familial hypercholesterolemia.2012; 23: 282–289.
10 Jiang L, Wu WF, Sun LY,. The use of targeted exome sequencing in genetic diagnosis of young patients with severe hypercholesterolemia.2016; 6: 36823.
11 Chiou KR, Charng MJ, Chang HM. Array-based resequencing for mutations causing familial hypercholesterolemia.2011; 216: 383–389.
12 Adzhubei IA, Schmidt S, Peshkin L,. A method and server for predicting damaging missense mutations.2010; 7: 248–249.
13 Leigh SE, Foster AH, Whittall RA,. Update and analysis of the University College London low density lipoprotein receptor familial hypercholesterolemia database.2008; 72: 485–498.
14 Tavtigian SV, Deffenbaugh AM, Yin L,. Comprehensive statistical study of 452 BRCA1 missense substitutions with classification of eight recurrent substitutions as neutral.2006; 43: 295–305.
15 Pengyu Su, Luya Wang, Jie Lin,. A novel mutation of the LDL receptor gene leading to familial hypercholesterolemia.2009; 111: 646–651.
16 Sun XM, Patel DD, Webb JC,. Familial hypercholesterolemia in China. Identification of mutations in the LDL-receptor gene that result in a receptor-negative phenotype.1994; 14: 85–94.
17 Mozas P, Castillo S, Tejedor D,. Molecular characterization of familial hypercholesterolemia in Spain: Identification of 39 novel and 77 recurrent mutations in LDLR.2004; 24: 187.
18 Bertolini S, Cassanelli S, Garuti R,. Analysis of LDL receptor gene mutations in Italian patients with homozygous familial hypercholesterolemia.1999; 19: 408–418.
19 Loux N, Saint-Jore B, Collod G,. Screening for new mutations in the LDL receptor gene in seven French familial hypercholesterolemia families by the single strand conformation polymorphism method.1992; 1: 325–332.
20 Zakharova FM, Damgaard D, Mandelshtam MY,. Familial hypercholesterolemia in ST-petersburg: The known and novel mutations found in the low density lipoprotein receptor gene in Russia.2005; 6: 6.
21 Genschel J1, Thomas HP, Kassner U,. Two novel LDL receptor mutations in familial hypercholesterolemia: C122Y and E296X.2001; 17: 354.
22 Schuster H1, Keller C, Wolfram G,. Ten LDL receptor mutants explain one third of familial hypercholesterolemia in a German sample.1995 15: 2176–2180.
23 Jiang L, Sun LY, Dai YF,. The distribution and characteristics of LDL receptor mutations in China: A systematic review.2015; 26; 5: 17272.
24 Nanchen D, Gencer B, Muller O,. Prognosis of patients with familial hypercholesterolemia after acute coronary syndromes.2016; 134: 698–709.
25 HovinghGK, DavidsonMH, KasteleinJJ,. Diagnosisandtreatmentof familial hypercholesterolaemia.2013; 34: 962–971.
26 Jelassi A, Guirim I, Najah M,. Limited mutational heterogeneity in the LDLR gene in familial hypercholesterolemia in Tunisia.2009; 203: 449–453.
27 Hattori H, Nagano M, Iwata F,. Identification of recurrent and novel mutations in the LDL receptor gene in Japanese familial hypercholesterolemia. Mutation in brief No. 248.1999; 14: 87.
28 Heath KE, Gudnason V, Humphries SE,. The type of mutation in the low density lipoprotein receptor gene influences the cholesterol-lowering response of the HMG-CoA reductase inhibitor simvastatin in patients with heterozygous familial hypercholesterolaemia.1999; 143: 41–54.
Yu-Jie ZHOU, MD, PhD, Department of Cardiology, Beijing Anzhen Hospital, Capital Medical University, 2 Anzhen Street, Chaoyang District, Beijing 100029, China; Lu-Ya WANG, PhD, Beijing Anzhen Hospital, Capital Medical University, Beijing Institute of Heart Lung and Blood Vessel Disease, The Key Laboratory of Remodeling-related Cardiovascular Disease, Ministry of Education, 2 Anzhen Street, Chaoyang District, Beijing 100029, China. E-mails: azzyj12@163.com (ZHOU YJ); wangluya@126.com (WANG LY).
October 16, 2017
February 4, 2018
March 20, 2018
June 28, 2018
杂志排行

Journal of Geriatric Cardiology的其它文章
- “Malignant” right coronary artery presenting as an ST-segment elevation myocardial infarction—a case report
- Influenza vaccination in acute coronary syndromes patients in Thailand: the cost-effectiveness analysis of the prevention for cardiovascular events and pneumonia
- The trend of change in catheter ablation versus antiarrhythmic drugs for the management of atrial fibrillation over time: a meta-analysis and meta-regression
- Early mortality and safety after transcatheter aortic valve replacement using the SAPIEN 3 in nonagenarians
- Depression and chronic heart failure in the elderly: an intriguing relationship
- CIED implantation in elderly patients: a single-center experience