Kennedy病患者电生理、病理学特点及临床误诊分析
2018-08-15康健捷,杨红军,邓兵梅等
肯尼迪病(Kennedy disease,KD)又称脊髓延髓型肌萎缩(spinal and bulbar muscular atrophy,SBMA)或迟发性脊髓延髓型肌肉萎缩,是一种少见的X连锁隐性遗传的神经元变性疾病。临床以缓慢进展的肌无力、肌萎缩、肌束震颤、延髓麻痹等下运动神经损害为主要表现,可伴有不同程度的感觉神经受累及雄激素不敏感综合征等表现[1,2]。基因检测是诊断KD的金标准,然而,因临床表现常不典型,极易被误诊为其他疾病,所以,患者在初诊时肌电图检查具有重要的诊断及鉴别诊断意义[3,4]。目前,我国对该病报道较少,尤其是电生理特点和病理学表现报道甚少。通过9例完整的肌电图和神经电图资料及1例肌肉病理资料并结合文献复习,分析KD的电生理特点、病理表现和临床误诊原因,为临床医师及神经电生理医师提供更深入的认识,以减少误诊误治。
1 资料与方法
1.1 研究对象 纳入2013 年1 月~2017 年9月广州军区广州总医院神经内科收治的9例KD 患者,经基因检测AR基因第一外显子CAG重复序列均> 40 个[4]。患者均为男性,年龄 41~59 岁,平均(49.11±6.25)岁,发病年龄31~48岁,从发病到基因确诊平均为(12.34±3.78) y。9例患者均无阳性家族史,均有肢体乏力,以近端受累为主。另选择20名与KD组年龄匹配的正常人作为正常对照组,均为男性;年龄40~60岁,平均年龄(50.4±7.05)岁;均排除有局部或广泛性的周围神经损害。
1.2 电生理检测方法 应用丹麦产Keypoint. 4肌电/诱发电位仪进行神经传导及针电极肌电图检测。神经传导检查包括:正中神经、尺神经、腓总神经、胫神经运动纤维的远端潜伏期(distal latency,DL)、复合肌肉动作电位波幅(compound muscle actionpotential,CMAP)及神经传导速度(motor conductionvelocity,MCV),正中神经、尺神经、腓肠神经感觉纤维的动作电位波幅(sensory nerveaction potential,SNAP)及神经传导速度(Sensoryconduction velocity,SCV),正中神经、尺神经和胫神经的F波出现率(F-Occurrenc),胫神经H反射潜伏期(H-reflex latency)。所有检查方法均遵循2008 年中华医学会神经电生理学组制定的肌电图规范化检测共识[5]。针极肌电图分别检查延髓(舌肌、胸锁乳突肌)、颈(三角肌、第一骨间肌)、胸(胸椎旁肌)、腰(股四头肌、胫前肌)4 个节段支配的肌肉,检测项目包括静息状态时自发电位、轻收缩时运动单位动作电位(motor unit action potential,MUAP)、重收缩时的募集相。神经源性损害表现为MUAP的时限增宽、波幅升高及多相波百分比增多,大力收缩时高波幅的单纯相或混合相;当合并自发电位出现正锐波、束颤电位、纤颤电位时为存在活动性的损害,否则为慢性损害[5]。患者皮温控制在30℃左右,室温控制在25 ℃左右。
1.3 肌肉活检 1例患者在超声引导下取双侧股四头肌各三条肌肉行肌肉活检病理检查。
2 结 果
2.1 电生理结果
2.1.1 神经电图 与正常对照组比较,KD组运动神经中腓总神经传导速度减慢,感觉神经中腓肠神经传导速度减慢,其余运动神经和感觉神经传导速度正常。正中神经、尺神经、腓总神经、胫神经复合肌肉动作电位(CMAP) 波幅降低(P<0.01),正中神经、尺神经、腓肠神经感觉神经动作电位(SNAP)波幅降低(P<0.01),以腓肠神经动作电位波幅异常率最高(88.89%)。正中神经和胫神经远端潜伏期延长(P<0.01)(见表2)。81.48% (44/54条)感觉神经动作电位波幅和76.38%(55/72条)运动神经动作电位波幅降低,8条的胫神经和10条腓总神经感觉动作电位未引出,KD组CMAP波幅为正常均值的53.79%~83.07%;77.78%(7/9例)的患者至少1根运动神经CMAP波幅降低;SNAP波幅明显降低,仅为正常均值的8.25%~55.98%;88.89%(8/9例)的患者至少有1条感觉神经的SNAP波幅降低;运动和感觉神经波幅均减低的占77.78%(7/9例)。正中神经、尺神经和胫神经的F波响应率分别为50.00%、77.78%和55.56%,正中神经和胫神经明显降低 (P<0.01)。9例KD病例中,仅3例(33.3%)胫神经可引出H反射,其潜伏期延长(P<0.01)。CMAP或SNAP波幅的异常率在4条运动神经和4条感觉神经之间,差异无统计学意义(P>0.05)。下肢腓肠神经SNAP波幅降低的程度较上肢正中神经为重,差异有统计学意义(P<0.01)。1例以咀嚼乏力起病的患者2005年和2010年神经电图均未见异常,2014年神经电图结果显示6条感觉神经动作电位波幅均明显降低,而4条运动神经动作电位均正常。
2.1.2 EMG 针电极检查结果 在9例患者的99块被检肌肉中,有纤颤正锐波和束颤电位等活动性损害改变的仅有12/99块(12.12%)。99块(100.0%)被检肌肉均有慢性神经源性损害,表现为高波幅长时限的运动单位动作电位(MUAP)伴募集减少。所有KD患者均表现为4个区域支配肌的同时累及 (见表2)。

表1 KD组和对照组的神经传导检查结果
注:*为两组所测神经传导结果对比,效率值<0.01;△为KD组腓肠神经SNAP波幅与正中神经SNAP波幅对比,效率值<0.01

表2 KD组患者不同区域肌肉神经源性损害情况[例(%)]
2.2 肌肉活检 股四头肌活检,HE染色可见肌纤维大小不等,萎缩纤维群组化分布,可见少量肥大肌纤维,部分肌纤维核内移,偶见坏死肌纤维伴少量炎症细胞浸润,未见变性及再生肌纤维。提示神经源性损害(见图1)。
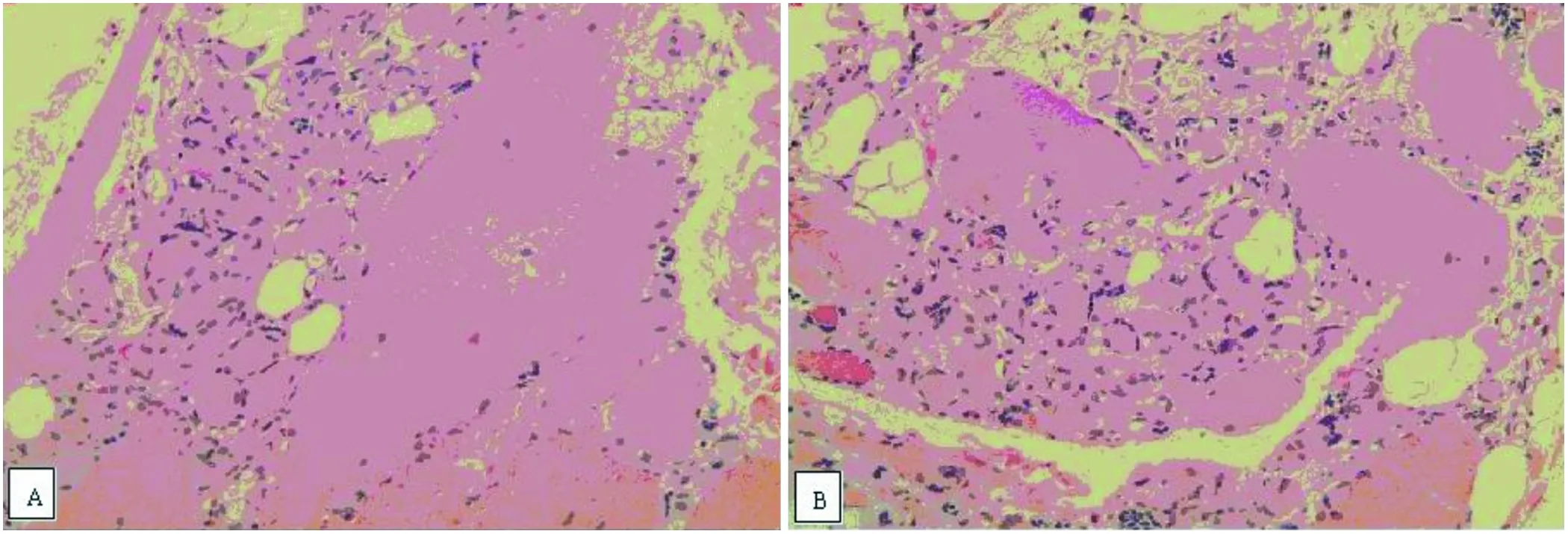
图1 A:肌纤维显著萎缩,萎缩纤维群组化分布,B:萎缩肌纤维形成多核肌巨细胞
3 讨 论
KD是一种少见的性连锁遗传病,X染色体长臂上的雄激素受体(androgen receptor,AR)基因的第一个外显子CAG重复序列异常扩增,导致其编码的AR多聚谷氨酰胺链延长,这种突变的雄激素受体与配体睾酮结合后由胞浆转入细胞核内,其毒性作用和降解异常等机制最终导致细胞变性坏死,而AR蛋白在脊髓和脑干运动神经元、初级和次级生殖器官、非生殖器官和骨骼肌均有表达,因此,临床出现神经、肌肉损害及男性特征减退。健康人该段CAG数目一般在11~35次,而在患者和女性携带者中该等位基因的CAG基因重复数一般在40~62次[4],>40个即可确诊。本研究的9例肯尼迪病患者CAG三核苷酸的重复序列数均超过40,均有肢体近端乏力,病程6~17 y,其中5例曾被误诊为运动神经元病,其余4例分别被误诊为进行性肌营养不良、线粒体肌病、多发性肌炎和重症肌无力。鉴于KD患者早期的误诊率高,既往文献对KD电生理学特征及病理学表现报道甚少,本研究对9例经基因诊断的KD患者电生理检测结果及1例肌肉病理检查结果进行分析,以加强对此病电生理学和病理学改变的认识并提高早期诊断率。
本研究结果显示运动神经中腓总神经传导速度减慢,感觉神经中腓肠神经传导速度减慢,其余运动神经和感觉神经传导速度均正常,提示神经髓鞘相对保持完整,与既往文献报道[6~8]一致。所测感觉和运动神经SNAP或CMAP波幅均显著下降。运动神经的损害程度在上下肢神经之间无明显差异,提示运动神经的损害不符合长度依赖的特点,而符合神经元损害的病变特点。腓肠神经动作电位波幅异常率最高(88.89%),而且腓肠神经SNAP波幅降低的程度较正中神经为重,提示下肢感觉神经损害程度重于上肢,与以往报道一致[6~8]。除1例病程为6 y的患者外,其余患者的腓肠神经SNAP波幅都在均值以下,其中8条的胫神经和10条腓肠神经感觉动作电位甚至未引出,支持KD患者下肢感觉神经损害较上肢严重,表现为长度依赖性,可能与除感觉神经元受损外,还存在感觉神经的轴索损害有关。KD病的细胞和动物模型研究也证实了存在轴浆运输障碍[9,10]。我们早期误诊为重症肌无力的1例患者,随访长达14 y,发病当年和发病5 y神经传导均未见异常,发病14 y电生理检查结果显示6条感觉神经动作电位波幅均明显降低,而4条运动神经动作电位均正常,提示感觉神经损害重于运动神经,可能损害发生也早于运动神经。既往研究发现[11],运动神经元核内具有AR染色活性的包涵体或聚集物的浓密聚集,而感觉神经元的聚集物散在分布于胞质中,这些研究结果提示感觉神经元和运动神经元损害的机制可能不同。因为KD患者临床上往往无肢体麻木的主诉,体格检查也无感觉系统阳性体征,所以感觉系统损害极易被忽视,从而易被误诊为只累及运动系统的运动神经元病,如本研究中有过半数患者曾在外院误诊为运动神经元病。因此,感觉神经动作电位有无缺失或波幅是否降低是KD与运动神经元病相鉴别的重要方面之一,值得高度重视,尤其是腓肠神经。不过,感觉神经传导正常不能除外KD,例如本研究中1例病程为6 y的患者感觉神经动作电位波幅均在正常低限,传导速度正常,运动神经动作电位均正常。以及1例长期随访的患者发病当年和发病5 y神经传导检查均未见异常。
根据文献报道[12],本研究针极肌电图检测的肌肉选择舌肌和双侧胸锁乳突肌、三角肌和第一骨间肌、胸段脊旁肌,股四头肌和胫骨前肌,分别代表了球、颈、胸、腰4 段部位。结果显示在被检肌肉中,仅有12.12%的肌肉有纤颤正锐波和束颤电位等活动性损害改变,而100.0%被检肌肉均表现为高波幅长时限的运动单位电位伴募集减少的慢性神经源性损害,提示KD患者球、颈、胸、腰4个区域支配肌同时受累。而运动神经元病则不同,其电生理异常随着疾病的发展而进展,针电极的失神经改变早期局限于起病区域,随着疾病的发展,病变区域逐渐扩大,最终累计球、颈、胸、腰4个区域的支配肌[12,13],表现为活动性改变和慢性再支配同时存在[14]。运动神经元病患者延髓支配的舌肌出现神经源性损害的时间早于脑桥三叉神经支配的咬肌或面神经支配的肌肉[15];而肯尼迪病患者的舌肌、咬肌和面神经支配肌均在疾病早期受累,部分患者早于肢体乏力的症状出现。本研究中77.78%(7/9)的患者在发病早期就出现舌肌萎缩和口周肌束震颤,1例患者发病早期出现咬肌无力,早于肢体乏力的症状出现。有文献报道少数患者可仅表现为咬肌无力和下颌下垂的症状[16,17]。由于该病以肢体近端无力起病,肌酸激酶普遍增高,早期易误诊为肌肉病。本研究中有3例患者被误诊为肌肉病。被误诊为多发性肌炎的患者病程6 y,临床肢体乏力症状轻微,而CK显著升高,高达2306U/L,运动神经及感觉神经传导检查结果均正常,在外院被误诊为多发性肌炎并给予激素冲击和免疫抑制剂治疗。在肌病和KD的鉴别诊断中肌电图至关重要,若肌电图检查显示为慢性神经源性损害及球、颈、胸、腰4个区域支配肌同时受累则对KD有提示作用。我们以往的研究发现[18]:患者CK水平与肌力下降水平以及病程均无关,提示患者的肌无力和运动功能下降并非主要由肌肉系统损害所致,CK不能作为判断病情和预后的因素,推测引起肌酶升高的原因可能是肌纤维直接被破坏和失神经支配引起肌肉萎缩所致。
文献报道,多数KD肌活检提示神经源性损害,本研究肌肉活检结果与此一致,可见肌纤维大小不等,萎缩纤维群组化分布,未见变性及再生肌纤维,提示神经源性损害。与肌电图检查结果相互印证。一般认为肌萎缩是运动神经损伤引起失神经支配的肌肉废用性萎缩,研究发现突变AR基因表达的多聚谷氨酰胺残基在肌纤维胞浆、核内聚集并形成包涵体,也可直接导致肌萎缩[19]。
因为KD临床表现常不典型,极易被误诊为运动神经元病、肌病,本研究中所有患者均有外院误诊误治情况。患者从发病到基因确诊时间较长,本研究为(12.34±3.78) y,而KD在男性人群患病率约为1/5万[20],临床对此病报道较少,因此,KD在中国还处于诊断不足的阶段。虽然KD在临床上和其他实验室检查中表现出较大的异质性,但其电生理改变高度一致,即球、颈、胸、腰4个区域支配肌同时受累,并表现为慢性神经源性损害,常伴感觉和运动神经动作电位波幅降低;感觉神经传导正常不能除外此病,腓肠神经波幅下降对KD有强烈提示作用。总之,对KD电生理和病理学特点加深认识有助于提高基因检测的针对性,节约医疗成本,提高疾病早期诊断率,减少误诊误治,缩短发病到基因确诊的时间。
