应用CRISPRi技术敲低耻垢分枝杆菌异柠檬酸脱氢酶基因
2018-08-14周凤竹胡亚文葛文雪张雪莲
周凤竹,胡亚文,葛文雪,张雪莲
复旦大学生命科学学院遗传工程国家重点实验室, 上海 200433
异柠檬酸脱氢酶(isocitrate dehydrogenase,ICD)是三羧酸(tricarboxylic acid cycle,TAC)循环中的关键酶,在辅酶存在时调节D-异柠檬酸氧化脱羧生成α-酮戊二酸和二氧化碳。该限速步骤是TCA循环中第1个生成NADPH的反应。结核分枝杆菌H37Rv菌株编码两种ICD,分别为ICD1和ICD2[1]。牛分枝杆菌卡介苗(bacillus Calmette-Guérin,BCG)菌株编码与H37Rv完全同源的ICD1和ICD2。利用工程菌表达的重组ICD1和ICD2在体外均有生物学功能[3],但它们对辅酶NADP的亲和力不同,Km(NADP)值及pH耐受性和热稳定性也有所不同[2]。在BCG菌体中敲除icd2无法检出ICD酶活,同时敲除株产生表型缺陷,需在培养基中添加谷氨酰胺才能正常生长,敲除icd1则对ICD酶活基本无影响,表明ICD2是BCG中负责碳通量进入TAC的关键酶,但ICD1的生理作用尚不清楚[4]。同时,由于敲除结核分枝杆菌中icd2的研究均没有成功,推测icd2可能是结核分枝杆菌生长的必需基因[5]。与BCG和结核分枝杆菌不同,耻垢分枝杆菌只编码1种ICD——MsICD,与结核分枝杆菌中ICD2高度同源,编码基因MSMEG_1654为耻垢分枝杆菌生长的必需基因[4]。
耻垢分枝杆菌由于生长较快,又同属分枝杆菌属,常被用来研究分枝杆菌的生理及代谢功能。由于分枝杆菌体内重组率非常低,其基因敲除一直是个难题。迄今虽然发展出许多方法来提高结核分枝杆菌的重组效率,如高滴度噬菌体特异性转导[6]、温敏型重组质粒介导的同源重组[7]等,但无法实现对必需基因的敲除。成簇的规律间隔的短回文重复序列干扰(clustered regularly interspaced short palindromic repeat interference,CRISPRi)技术是一种基于Cas9的调控基因表达的技术,其中Cas9是来自Ⅱ型CRISPR系统的RNA介导的DNA内切核酸酶[8]。当这种缺乏核酸内切酶活性即无催化功能的Cas9与设计好的向导RNA共表达时,可产生一种DNA识别复合物,这种复合物可特异性干扰RNA聚合酶或转录因子与DNA的结合,从而阻碍DNA转录[9-11]。Rock等对这种技术改进,使其能应用于分枝杆菌属的细菌转录干扰[12]。本研究以耻垢分枝杆菌中编码ICD的MSMEG_1654基因为靶基因,利用CRISPRi技术,成功构建MsICD敲低菌株,在无水四环素(anhydrotetracycline,ATc)诱导下表达dCas9和单向导RNA(single guide RNA,sgRNA),对其进行干扰,在细菌基因组不发生变化的情况下MsICD的转录和表达水平下降,为构建必需基因敲除提供了可靠方法,并为后期研究结核分枝杆菌ICD敲低及分枝杆菌ICD在碳源代谢和碳通量分流调控机制中的作用提供了基础。
1 材料与方法
1.1 材料
1.1.1引物表1中的引物全部由生工生物工程(上海)股份有限公司合成。
表1本研究使用的引物
Tab.1Primersusedinthisstudy

PrimerSe-quencesgRNA-S5′-GG-GAGCGAATTTCCGTACCACCGG-3′sgRNA-A5′-AAAC-CCGGTGGTACGGAAATTCGC-3′ICDq-S5′-GCAGC-CGACCATCATCTACA-3′ICDq-A5′-AC-CGAGATGTCGCTGGTTTT-3′sigA-S5′-CGTC-CGGCGACTTCGTGT-3′sigA-A5′-CCT-TGCCGATCTGCTTGAGGTA-3′
1.1.2其他材料耻垢分枝杆菌mc2155菌株、大肠埃希菌DH5α菌株由本实验室保存。PLJR962质粒由哈佛大学Sarah M. Fortune课题组惠赠。抗结核分枝杆菌ICD2兔多克隆抗体、抗结核分枝杆菌DosR兔多克隆抗体由吉尔生化(上海)有限公司定制,辣根过氧化物酶标记的羊抗兔IgG多克隆抗体购自Proteintech。主要试剂包括BsmBⅠ(NEB)、T4连接酶(NEB)、无水四环素(TaKaRa)、PrimeScript RT Reagent Kit with gDNA Eraser(TaKaRa)、2×T5 Fast qPCR Mix(SYBR Green I)(北京擎科新业生物技术有限公司)、Middlebrook 7H9 Broth(Difco)等。其他化学试剂均购自生工生物工程(上海)股份有限公司。
1.2 方法
1.2.1敲低菌株的构建将合成的sgRNA-S和sgRNA-A按1∶1摩尔比加入去离子水中退火,95 ℃ 2 min,自然冷却至室温。将PLJR962载体及退火后的sgRNA分别在55 ℃用BsmBⅠ酶切4 h,回收产物后,以T4连接酶室温连接2 h,将连接产物转化DH5α感受态细胞并涂布于卡那霉素抗性平板,筛选阳性克隆。测序验证载体构建成功后,将阳性质粒电转入耻垢分枝杆菌mc2155感受态细胞,并涂布于卡那霉素抗性平板,筛选阳性克隆菌落。
1.2.2细菌生长差异挑选阳性克隆菌落,扩大培养。细菌生长至OD600为 0.6 后,离心收集菌体,并加入培养基重悬菌体。将菌液密度梯度稀释至10-5,每个梯度取3 μL,分别点板于含有30 μg/mL卡那霉素的7H10-OADC固体板及含30 μg/mL卡那霉素、100 μg/mL无水四环素的7H10-OADC固体板。取上述重悬后的菌液,加入50 mL 7H9-OADC液体培养基(含30 μg/mL卡那霉素和100 μg/mL无水四环素),使初始OD600为 0.02,37 ℃ 100 r/min振荡培养,于接种后0、4、8、12、24、30、36、48、72 h取样检测OD600。每次实验设置3个平行重复。
1.2.3icd转录水平检测将阳性菌落转移至液体培养基中扩大培养,至OD600为 0.3,加入100 μg/mL无水四环素,继续37 ℃ 100 r/min培养14 h,室温 4 000 r/min离心10 min,收集菌体。采用TRIzol法抽提细菌RNA[13]。去除基因组DNA的反转录采用PrimeScript RT Reagent Kit with gDNA Eraser试剂盒,程序为37 ℃ 15 min,85 ℃ 5 s。取100 ng反转录产物进行定量聚合酶链反应(quantitative polymerase chain reaction,qPCR),程序为95 ℃ 1 min,95 ℃ 10 s,60 ℃ 5 s,72 ℃ 15 s,共40个循环,72 ℃采集荧光信号。以sigA为内标,设置3个平行组,按目标基因相对于内标基因的扩增量为2-ΔΔCt处理数据,其中-ΔΔCt=-(ΔCt,q-ΔCt,cb),ΔCt,q为目标基因循环数,ΔCt,cb为内标基因循环数。
1.2.4ICD蛋白表达水平检测将阳性菌落转移至液体培养基(含或不含100 μg/mL无水四环素)中培养至OD600为0.6,室温 4 000 r/min离心10 min,收集菌体,用磷酸盐缓冲液(phosphate buffered saline,PBS)洗2次,收集菌体。加入适量1×PBS重悬,以100 W,开2 s,关2 s,循环100次,冰水浴超声破碎细菌,13 000 r/min离心30 min。取上清液,BCA法测定菌体蛋白浓度。取50 μg全菌蛋白进行蛋白免疫印迹检测,一抗为抗结核分枝杆菌ICD2兔多克隆抗体,以抗结核分枝杆菌DosR兔多克隆抗体为对照,二抗为辣根过氧化物酶标记的羊抗兔IgG多克隆抗体。
1.2.5ICD酶活测定ICD酶活测定体系[3]包括20 mmol/L三乙醇胺缓冲液(pH 7.5)、2 mmol/L NADP、10 mmol/L MgCl2、100 mmol/L NaCl、50 μg全菌蛋白,用ddH2O补齐至180 μL,最后加入 3 mmol/L异柠檬酸至终体积为200 μL,立即置入酶标仪测定OD340,25 ℃连续读数15 min。根据Unit=ΔOD340/(ε340·l·Δtm)计算比活力。其中ΔOD340为最大反应速度期间OD340值之差,ε340=6.22 mmol/(L.cm),l=0.45 cm。设置3个平行组。
2 结果
2.1 敲低菌株的构建
本研究使用的CRISPRi技术是通过无水四环素诱导表达PLJR962质粒中的dCas9和sgRNA,后两者同时结合特异DNA位点,阻止转录的起始或延伸。dCas9来自嗜热链球菌,可结合sgRNA,但无催化活性。采用这种CRISPRi技术时,需设计用来靶定特异DNA位点的sgRNA。前间区序列邻近基序(protospacer adjacent motif,PAM)是一段较短的DNA序列,可促使Cas9-sgRNA复合体形成R-环并阻碍转录。本研究采用的PAM序列为5′-CTTCT-3′,位于靶定基因MSMEG_1654的5′端附近。
将构建完成的质粒电转入耻垢分枝杆菌后,sgRNA与dCas9结合,sgRNA识别互补配对的基因组中的碱基序列并结合。转录开始后,RNA聚合酶到达sgRNA结合位点,由于此处DNA链被dCas9和sgRNA结合无法打开,即无法继续进行转录,从而达到降低表达水平的目的。
2.2 细菌生长差异
经过抗性筛选后,将生长至相同密度的菌液梯度稀释后点板,在培养基中添加无水四环素诱导转录抑制时,ICD-KD菌株的生长速率明显减缓(图 1),在10-2(约2×105个细胞/mL)已产生肉眼可见的生长差异。随着点板菌液密度降低,敲低菌株的生长减缓现象愈加明显。液体培养基中的生长曲线(图 2)也表明,ICD-KD菌株在添加无水四环素条件下生长速率显著减缓,并在对数期生长速率差异明显。

The density of bacteria is 10-5to 10-1, approximately 2×102to 2×106cells/mL. 3 μL bacteria at different density were added into every grid.
图1敲低菌株在固体培养基中生长减缓
Fig.1GrowthofICD-KDstrainsinsolidmedium

The initialOD600of bacteria is about 0.02. At log phase, the growth rate of ICD-KD strains was lower than vector strains.
图2敲低菌株在液体培养基中生长减缓
Fig.2GrowthofICD-KDstrainsinliquidmedium
2.3 icd转录水平检测
在无水四环素诱导下,敲低成功的ICD-KD菌体内目的基因转录水平降低。同样条件下培养细菌,抽提等量菌体内RNA检测目的基因转录水平(图 3)。结果显示,与转入空载的耻垢分枝杆菌相比,ICD-KD菌株在加入无水四环素诱导后MSMEG_1654基因转录水平显著降低(P<0.001),表明菌体内icd转录受到影响。
2.4 ICD蛋白表达水平检测
在液体培养基中培养细菌,约24 h后达对数期。将此时的菌体超声破碎离心,取上清液测定总蛋白浓度,检测ICD蛋白表达水平(图 4)。上样量为50 μg时,全菌蛋白的十二烷基硫酸钠-聚丙烯酰胺凝胶电泳(sodium dodecyl sulfate-polyacrylamide gel electrophoresis, SDS-PAGE)条带基本一致,无明显差异。蛋白免疫印迹检测结果表明,在无水四环素诱导下,与转入空载的菌株相比,ICD-KD菌株中ICD表达水平显著降低。

The transcriptional level ofMSMEG_1654 in ICD-KD strains cultured with ATc was significantly decreased compared to other strains.
图3敲低菌株的MSMEG_1654转录水平
Fig.3RelativetranscriptionallevelofMSMEG_1654inICD-KDstrains
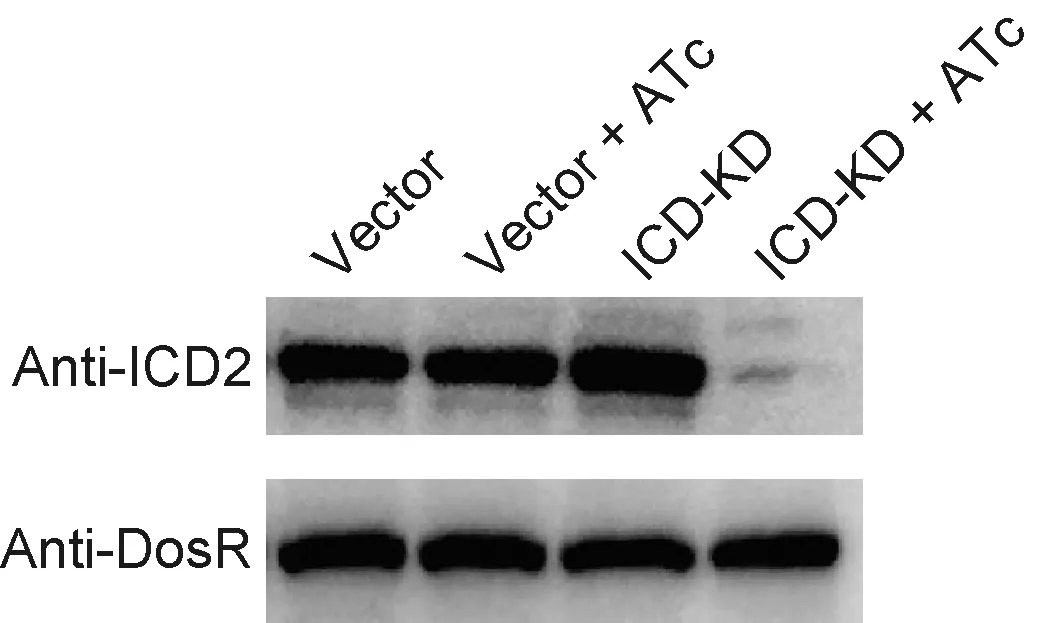
The anti-ICD polyclonal antibody was used in Western blotting. ICD expression in ICD-KD strains was significantly decreased compared to wild-type strains and ICD-KD strains without ATc. DosR was used as an internal reference. The molecular weight of ICD protein is about 86.2 kDa, and that of DosR is about 23 kDa.
图4敲低菌株的ICD蛋白表达水平
Fig.4ProteinexpressionofICDinICD-KDstrains
2.5 ICD酶活测定
取2.4中不同培养条件获得的全菌蛋白,检测ICD酶活水平(图 5)。在无水四环素存在时,ICD-KD菌株的全菌ICD酶活水平显著降低(P<0.000 1),与ICD转录水平和蛋白水平检测结果相符。
综合上述结果,表明耻垢分枝杆菌ICD-KD菌株构建成功,ICD低转录、低表达,酶活也受到抑制,严重影响耻垢分枝杆菌的生长繁殖,与野生型细菌相比,ICD-KD菌株生长缓慢。
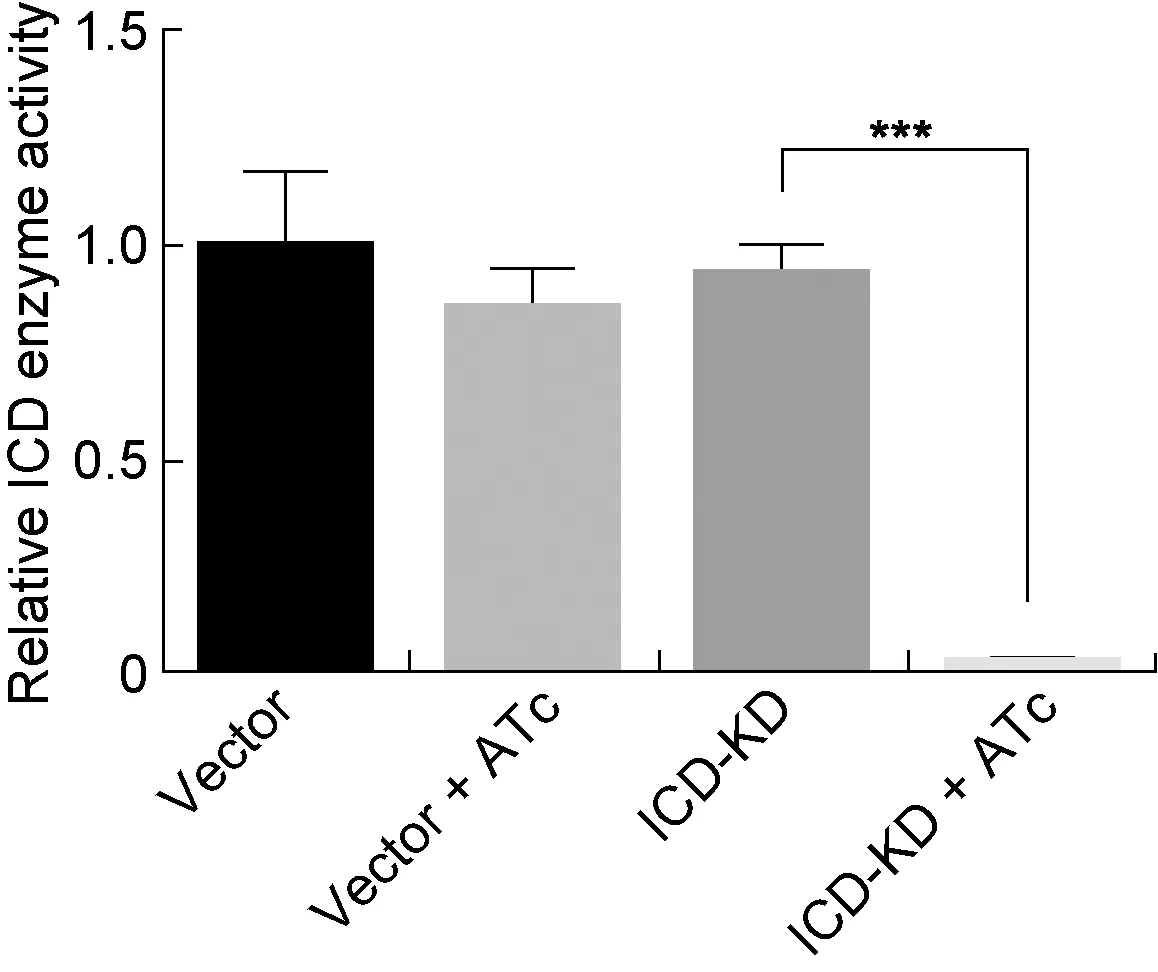
ICD enzyme activity in ICD-KD strains cultured in medium containing ATc was decreased compared to ICD-KD strains without ATc.
图5敲低菌株的相对ICD酶活水平
Fig.5RelativeICDenzymeactivityinICD-KDstrains
3 讨论
目前,基因功能研究方法包括启动子置换和诱导型蛋白降解系统,可使目的基因表达水平发生改变[14-16],但可能需几个月才能靶向单个基因。转座子测序(transposon sequencing, Tn-seq)允许同时评估数十万个功能丧失突变体,但在结核分枝杆菌中不能做到人为调控表达水平,无法操纵重要基因[5]。此外,这些方法不能提供简单的机制来同时调节多个基因以阐明遗传相互作用[9]。对分枝杆菌基因功能的研究主要依赖目的基因敲除,但分枝杆菌菌体内缺少重组酶,同源重组率非常低,基因敲除难度相对较大,特别是对必需基因的功能研究更是一个瓶颈。本研究使用CRISPRi技术,非常简单方便,只需构建含有特异sgRNA的单一质粒(sgRNA骨架)并转化入分枝杆菌[17]。sgRNA骨架的构建也十分快捷。CRISPRi是诱导型转录干扰,可操作一些比较重要的基因,不受必需基因的限制。相比直接基因敲除,其更可控[18-20]。通过改变靶定的PAM区,可调节CRISPRi对转录的抑制水平,从而改变基因敲低的幅度。但CRISPRi存在一些不足,如极性效应:dCas9靶定位点下游与靶定基因处于同一操纵子内的所有基因转录都将被抑制[9]。
ICD是TAC中的关键限速酶。碳通量通过其底物异柠檬酸进入代谢通路,是分枝杆菌脂肪酸代谢的重要途径。已有研究表明,结核分枝杆菌的碳代谢过程对其致病能力起重要作用[21]。在代谢不同碳源时,结核分枝杆菌和耻垢分枝杆菌的ICD酶活水平发生改变[4]。研究分枝杆菌代谢碳源的机制有利于新型抗结核药物的开发。有研究发现,在耻垢分枝杆菌中敲除MSMEG_1654会导致细菌发生谷氨酰胺表型缺陷,即无法在没有添加谷氨酰胺的培养基中生存[4]。因此,本研究采用CRISPRi技术使细菌不产生表型缺陷,并能适当降低目的基因表达水平[22]。
本研究中,MSMEG_1654基因与上下游基因均不位于同一操纵子,因此利用CRISPRi的转录抑制可发挥稳定作用,不必考虑极性效应的影响。耻垢分枝杆菌中MSMEG_1654敲低后,基因转录水平大幅降低,蛋白表达水平也明显下降, ICD酶活显著降低。MSMEG_1654是必需基因,表达被抑制时会对细菌生长造成影响。因此,在固体培养基中添加无水四环素诱导转录抑制时,敲低菌株的生长明显减慢。必需基因敲低后会明显影响细菌生长,这也是应用CRISPRi技术敲低必需基因的显著特征。后续研究将继续探索ICD在TAC及碳源代谢中的具体功能和机制,如细菌在代谢不同碳源过程中ICD的作用,并进一步分析分枝杆菌ICD在碳源代谢和碳通量分流调控中的作用。本研究利用CRISPRi技术快速敲低耻垢分枝杆菌必需基因的方法,也为后续结核分枝杆菌必需基因的敲低和功能研究开展提供了重要基础。
