玉米雄穗分枝数主效QTL定位及qTBN5近等基因系构建
2018-08-10代资举王新涛张莹莹席章营李保全
代资举 王新涛 杨 青 王 艳 张莹莹 席章营 李保全,*
玉米雄穗分枝数主效QTL定位及近等基因系构建
代资举1王新涛1杨 青1王 艳1张莹莹1席章营2李保全1,*
1河南省农业科学院作物设计中心, 河南郑州 450002;2河南农业大学农学院 / 河南省粮食作物协同创新中心, 河南郑州 450002
立足于发掘玉米雄穗分枝数优异基因资源, 利用郑单958骨干亲本郑58和昌7-2构建的188个重组自交系(recombinant inbred line, RIL)家系群体, 结合288个多态性分子标记构建的连锁图谱和2年玉米雄穗分枝数表型数据, 运用完备复合区间作图法进行QTL定位, 共检测到5个控制玉米雄穗分枝数的一致性主效QTL, 分别位于玉米5条染色体上。通过连续回交及分子标记辅助选择构建了位于5.05的控制雄穗分枝数主效QTL-近等基因系(near isogenic line, NIL), 对基因遗传效应进行了验证, 并将进一步定位在13.2 Mb区间之内, 为玉米雄穗分枝数主效基因的精细定位及分子育种奠定基础。
玉米; 雄穗分枝数; QTL定位; 近等基因系
玉米雄穗分枝数(tassel branch number, TBN)作为衡量雄穗大小的重要指标, 是玉米育种与种子生产过程中被研究的与产量形成有关的重要农艺性状之一。Geraldi等[1]的研究表明雄穗分枝数与产量呈负相关。然而在玉米杂交种种子生产过程中却要求作父本的亲本植株具有较为发达的雄穗, 以利于提高制种质量与降低成本[2]。同时较小适中的雄穗又是理想株型的构成要素之一, 过度发达的雄穗影响株型的冠层结构[3]。因此在育种实践中选育适度减少雄穗分枝数的杂交种是目前玉米育种的一个趋势, 而研究玉米雄穗分枝数基因的遗传效应, 挖掘玉米雄穗分枝数基因资源可以为玉米产量和株型相关性状的遗传改良提供依据。
随着现代分子生物技术的发展, 一些控制玉米雄穗分枝数QTL被鉴定出来。Berke和Rocheford[4]利用F2群体定位到3个雄穗分枝数目QTL, 分布于第2、第4、第7染色体上, 其中第7染色体上的QTL贡献率达35.3%。Mickolson等[5]利用重组自交系群体, 定位到6个雄穗分枝数目QTL, 分布于第1、第2、第3、第4染色体上, 其中第2染色体QTL贡献率达19.2%。汤华等[6]利用N87-1×综3构建F2:3群体, 定位到9个雄穗分枝数目QTL, 分布于第1、第3、第4、第5、第9、第10染色体上, 贡献率为6.11%~12.01%。Upadyayula等[7]利用BC1S1家系群体, 定位到2个雄穗分枝数目QTL, 分布于第4、第7染色体, 贡献率分别为7.9%和11.7%。高世斌等[8]利用N87-1×9526构建F2:3群体, 定位到6个雄穗分枝数目QTL, 分布于第2、第4、第7、第10染色体上, 贡献率为5.6%~28.4%。王迪等[9]利用齐319×黄早四和披478×黄早四分别构建了2套F2:3群体, 定位到40个雄穗分枝数目QTL, 10条染色体上均有分布, 贡献率为2.93%~23.78%。Brown等[10]利用5000份重组自交系群体组成的(nested association mapping, NAM)群体, 检测到39个雄穗分枝数目QTL。杨钊钊等[11]以黄早四为共同亲本构建了11个不同组合的重组自交系群体, 定位到11个雄穗分枝数目QTL, 分布于第2、第3、第4、第5、第7、第8染色体, 贡献率为9.7%~20.9%。Chen等[12]利用F2群体和高密度遗传图谱检测到7个控制雄穗分枝数QTL, 分布于第1、第3、第4、第5、第7、第8、第9染色体上。Wu等[13]利用NAM群体和全基因组关联分析检测到63个控制雄穗分枝数QTL。而Xu等[14]利用栽培玉米和类蜀黍(teosinte)发展作图群体, 利用SNP标记全基因组扫描检测到14个控制玉米雄穗分枝数QTL。以上研究表明玉米雄穗分枝数是多基因控制数量性状, 控制该性状基因在多数染色体上都是存在的, 且存在主效QTL。但由于不同的研究所用到的F2:3、RIL、BC1S1等群体类型易受到遗传背景的影响及试验环境和标记密度等因素干扰, 使得彼此定位结果存在差异, 具有一致性且贡献率较大的QTL比较少, 因此难以通过精细定位克隆雄穗分枝数基因。
玉米雄穗分枝发育涉及众多关键基因, 基于突变体的遗传分析, 已鉴定分离了多个调控玉米雄穗分枝数发育基因, 如参与玉米花序分枝分生组织调控的基因突变体系列()、()和(), 主要表现为雄穗基部分枝数增多, 其中和基因分别位于第7染色体的两个不同区域,编码含有一个Cys2-His2锌指结构域且具植物特异性的类EPF蛋白, 而基因编码6-磷酸海藻糖酶用以修饰进入花序分生组织的糖信号, 进而调控的转录活性,基因位于第3染色体, 是一个含有LOB-domain的转录因子, 在茎顶端分生组织和花序分生组织细胞中表达较高, 其中起关键作用, 调节和表达[15-17]。()突变体雄穗只有一个主轴而没有分枝、小穗和小花, 导致突变的原因是位于第3染色体基因编码起始位点上游存在转座子插入, 该基因编码一个碱性螺旋-环-螺旋(helix-loop-helix)结构蛋白, 是一个植物特有的bHLH 转录因子[18]。()主要控制玉米叶舌和叶耳的发育, 也参与花序建成的调控, 雄穗下部长分枝不能启始, 该基因位于第3染色体上, 编码一个bZIP结构蛋白[19]。()基因是位于第1染色体上的一个编码丝氨酸/苏氨酸激酶, 参与生长素极性运输,突变体表现为雄穗分枝难以分化、小穗分生组织减少[20]。而()和()基因分别位于第1和第4染色体, 是一类转录因子, 增强其表达可以增加玉米雄穗分枝和花药数[21]。尽管这些突变体导致了雄穗分枝数的改变, 为雄穗分枝数相关基因的克隆及功能研究提供了参考, 但部分突变体表现出其他性状(如雌穗性状、植株形态性状等)的改变, 影响了优异种质材料的育种利用。
本研究首先利用郑单958的骨干亲本郑58和昌7-2构建的重组自交系群体对控制玉米雄穗分枝数主效QTL进行鉴定, 然后构建主效QTL的近等基因系验证基因遗传效应, 开发连锁标记, 利用重组株系缩短定位区间, 为玉米雄穗分枝数主效基因的精细定位和克隆及分子育种实践奠定基础。
1 材料与方法
1.1 供试材料与田间试验
以生产上大量应用玉米品种郑单958的2个优良自交系亲本郑58和昌7-2杂交, 采用单粒传法连续自交至F7:8构建188个重组自交系群体。分别于2015年和2016年夏季在河南省农业科学院原阳试验基地种植188个重组自交系及亲本, 采用随机区组设计, 不设重复, 单行区, 行长2 m, 每行10穴, 每穴1株, 植株授粉15 d后, 调查雄穗分枝数, 每行从第2株开始, 连续调查5株完整雄穗, 统计各家系的雄穗分枝数平均值用于进一步分析。-NILs构建试验材料于2008—2017年夏季种植于河南郑州, 冬季种植于海南三亚, 春季在河南省农业科学院原阳试验基地温室加代。
1.2 分子标记检测
按CTAB法从玉米幼苗叶片组织提取分离基因组DNA。引物合成的序列来自于玉米基因组数据库(MaizeGDB, http://www.maizegdb.org/)。PCR扩增体系为10 μL, 包括DNA模板2.0 μL、5 U µL–1DNA聚合酶0.1 μL、10×buffer 1.6 μL、10 mmol L–1dNTP 0.15 μL、Primer (M13+F +R) 0.4 μL和ddH2O 5.75 μL。扩增程序为94°C 预变性5 min; 94°C变性60 s, 60°C退火50 s, 72°C延伸50 s, 35个循环; 72°C延伸10 min, 4°C保存。电泳检测体系为2 μL PCR产物、7 μL甲酰胺和0.04 μL liz 500分子量内标(Applied Biosystems, USA)。95°C变性3 min后, 在ABI 3500XL基因分析仪进行电泳。用Genographer 2.1软件分析带型数据。
1.3 玉米雄穗分枝数QTL定位
从1928个分子标记中筛选到在郑58与昌7-2之间有多态性的分子标记300个, 用于植株的基因型鉴定。经c2检验后, 选择扩增位点符合孟德尔分离定律(1∶1)的引物288个, 利用MapMaker/EXP 3.0 软件构建群体的遗传图谱[22], 采用Haldane函数, 图距单位为cM (centiMorgan)。根据引物序列比对B73 RefGen-v3基因组序列, 找出标记在图谱上的物理位置, 利用MapChart 2.1绘制物理图谱。利用188个家系的雄穗分枝数平均数进行QTL定位, 选用QTL IciMapping Version 4.0软件的复合区间作图法[23]分析QTL效应。设定QTL检测的LOD阈值为3.0, PIN值为0.001。
1.4 主效QTL-qTBN5近等基因系构建
利用分子标记辅助选择(marker-assisted selection, MAS)及表型选择创制-NILs流程(图1)。在-NILs创制过程中, 子代群体只保留经分子标记选择基因型正确、雄穗分枝数与受体亲本植株表型具有显著差异、而其他性状与轮回亲本相近的单株, 并严格进行杂交或自交授粉。以昌7-2为供体亲本, 郑58为受体材料并作为轮回亲本(recurrent parent)回交5代后自交, 用140个多态性SSR进行全基因组背景选择, 构建以郑58为受体的染色体片段代换系(chromosome segment substitution line, CSSL)。然后利用连锁标记umc1853选择基因型为且雄穗分枝数与受体亲本存在显著差异的CSSL, 用均匀分布于全基因组的288个多态性标记进行背景分析, 选出片段较少且携带目标基因片段的CSSL与郑58杂交、回交, 最后自交, 选择基因型纯合且除雄穗分枝数外其他性状没有明显差异的植株, 表型稳定遗传后创制- NILs。

图1 qTBN5-NILs的构建流程
2 结果与分析
2.1 亲本与RIL家系雄穗分枝数表型分析
对亲本郑58和昌7-2及188个RIL家系的两年雄穗分枝数调查结果(表1)表明, 188个RIL家系雄穗分枝数的总平均数分别为10.7个和11.2个, 家系间从最少的2.4个和2.8个到最多的22.6个和21.4个, 变异系数分别为43.60和38.83, 分布范围较广。而且广义遗传率(2)较高, 为89.74%, 表明RIL家系群体大部分表型变异受遗传控制。
分别以188个RIL家系雄穗分枝数平均值的两年数据制作频次分布, 呈现为一种连续的近似正态分布(图2), 表明玉米雄穗分枝数是典型的多基因控制的数量性状。亲本郑58雄穗分枝数的平均个数2年分别为4.6个和4.4个, 而昌7-2两年分别为17.8个和17.2个, 多数家系雄穗分枝数的平均值都介于两个亲本之间, 但也有部分家系雄穗分枝数在两个亲本区间之外, 说明控制雄穗分枝数基因存在双向重组超亲分离, 双亲均含有影响雄穗分枝数性状的增效、减效基因。
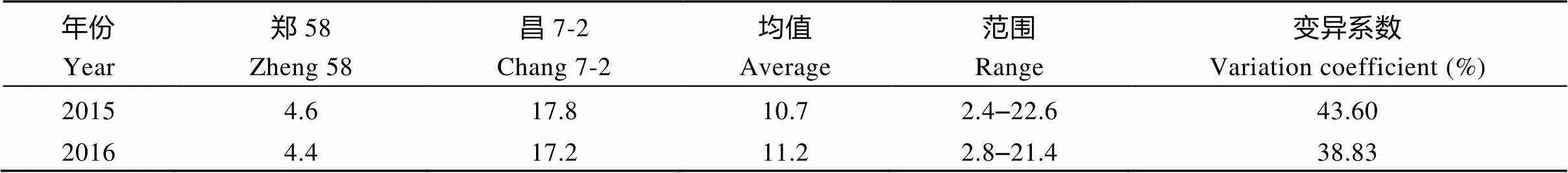
表1 亲本及RIL家系在两年环境下的雄穗分枝数的表型分析
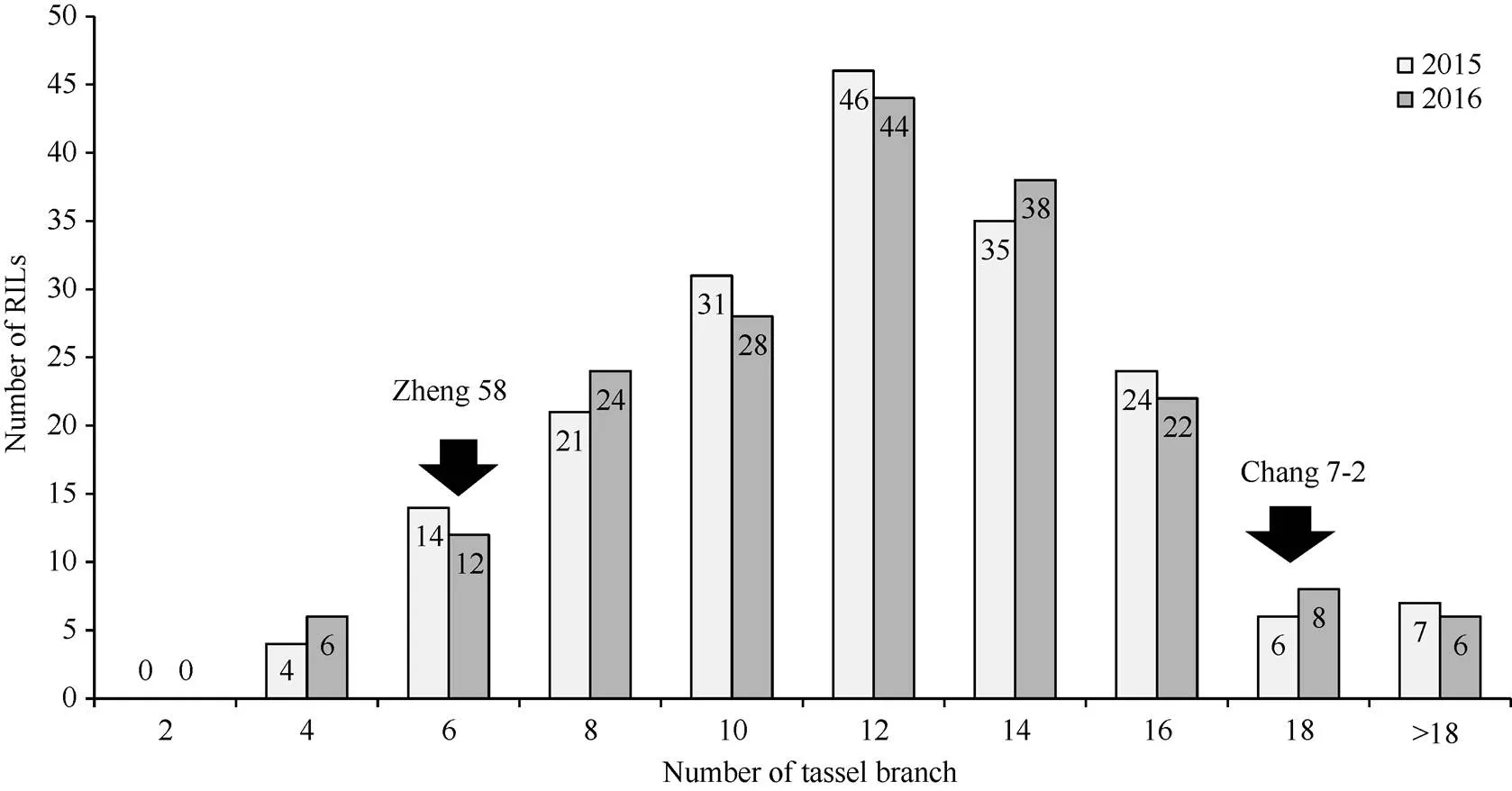
图2 RIL家系雄穗分枝数频次分布
2.2 QTL检测与遗传效应分析
连锁图谱共包含288个在郑58和昌7-2之间存在多态性的分子标记, 覆盖玉米整个10条染色体的基因组, 除第4和第7染色体存在2个较大gap外, 其他标记分布较为均匀。根据引物序列参考比对B73 RefGen-v3基因组序列, 明确了分子标记在图谱上的物理位置(图3)。
利用188个RIL家系两年的雄穗分枝数表型数据, 经复合区间作图法在包含288个分子标记的连锁图谱上一共检测到了5个两年均可重复检测的主效QTL, 分别位于第3、第4、第5、第7和第10染色体上, 即、、、和(图3), 各QTL的贡献率变异范围在5.81%~ 19.92%之间(表2), 在5个QTL中贡献率超过10%的有3个, 最高的是位于第5染色体5.05上的。在被检测到的5个QTL中, 有3个加性效应值为负值, 表明来自于郑58的等位基因可以使雄穗分枝数减少; 而加性效应值为正值、位于第3和第5染色体上的和的贡献率较高, 分别达到了13.67%和19.92%, 表明来自于亲本昌7-2的增效等位基因可以使雄穗分枝数显著增加, 这与作为父本的昌7-2自身具有相对较多的雄穗分枝数有关。
2.3 qTBN5-NILs的表型分析
根据雄穗分枝数主效QTL-定位(图4-a), 利用回交、自交和分子标记前景及背景选择, 成功创制了-NILs。图4-b是-NILs基因型, 除位于第5染色体上基因所在片段来自昌7-2之外, 其他背景均来自郑58。对创制成功的-NILs及其受体亲本郑58和供体亲本昌7-2的雄穗分枝数、株高、穗位高、抽雄期和雄穗主轴长进行调查,检验分析表明: 受体亲本郑58和供体亲本昌7-2的雄穗分枝数与-NILs之间均存在显著性差异(图4-c), 而其他主要性状如株高、穗位高、抽雄期和雄穗主轴长在郑58与-NILs之间差异不显著(图4-d~g)。由此表明,基因的导入可以使郑58雄穗分枝数显著增加, 但是达不到昌7-2水平, 而且并没有造成郑58其他主要性状的改变。
2.4 雄穗分枝数主效QTL-qTBN5的进一步定位及连锁标记开发
根据在第5染色体5.05的定位区间, 利用CSSL和郑58发展的BC1F2作图群体, 提取DNA筛选交换单株, 种植重组单株自交发展株系, 纯合株系用于表型考察和进一步定位(图5-a), 从网上公布的引物数据库及相关序列, 开发筛选到3个与紧密连锁的分子标记, 即引物phi333597、e2040、e2042 (图5-b), 结合重组株表型将进一步定位在phi333597和e2042的13.2 Mb区域之内, 为的精细定位和分子标记辅助育种奠定基础。

图3 雄穗分枝数性状主效QTL在标记连锁图上的位置
连锁图上左边数字表示标记在B73 RefGen-v3基因组序列图谱上的物理位置。
Physical map distances (Mb) referring B73 RefGen-v3 are shown on the left of linkage map.
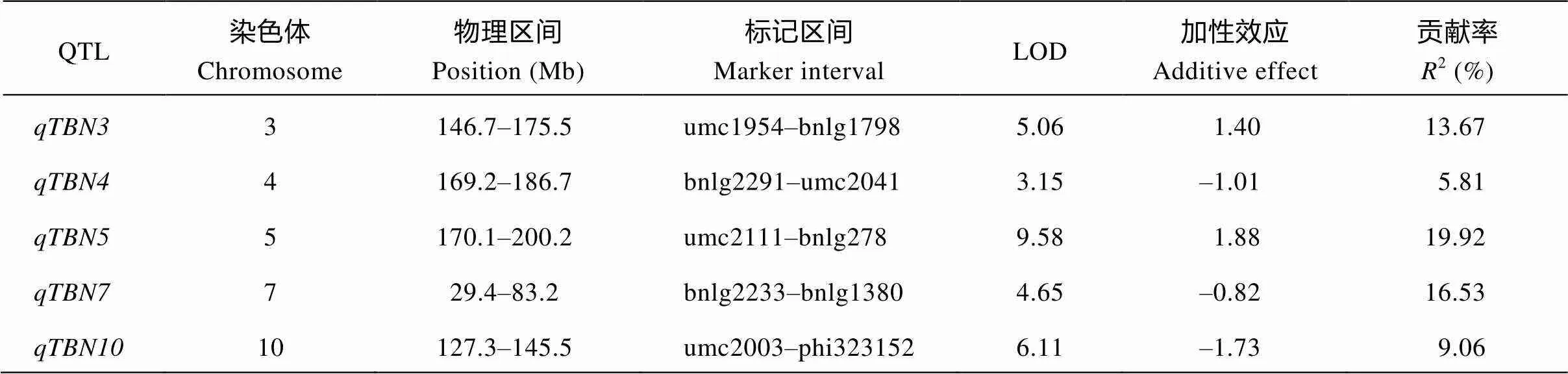
表2 玉米雄穗分枝数主效QTL及其遗传效应
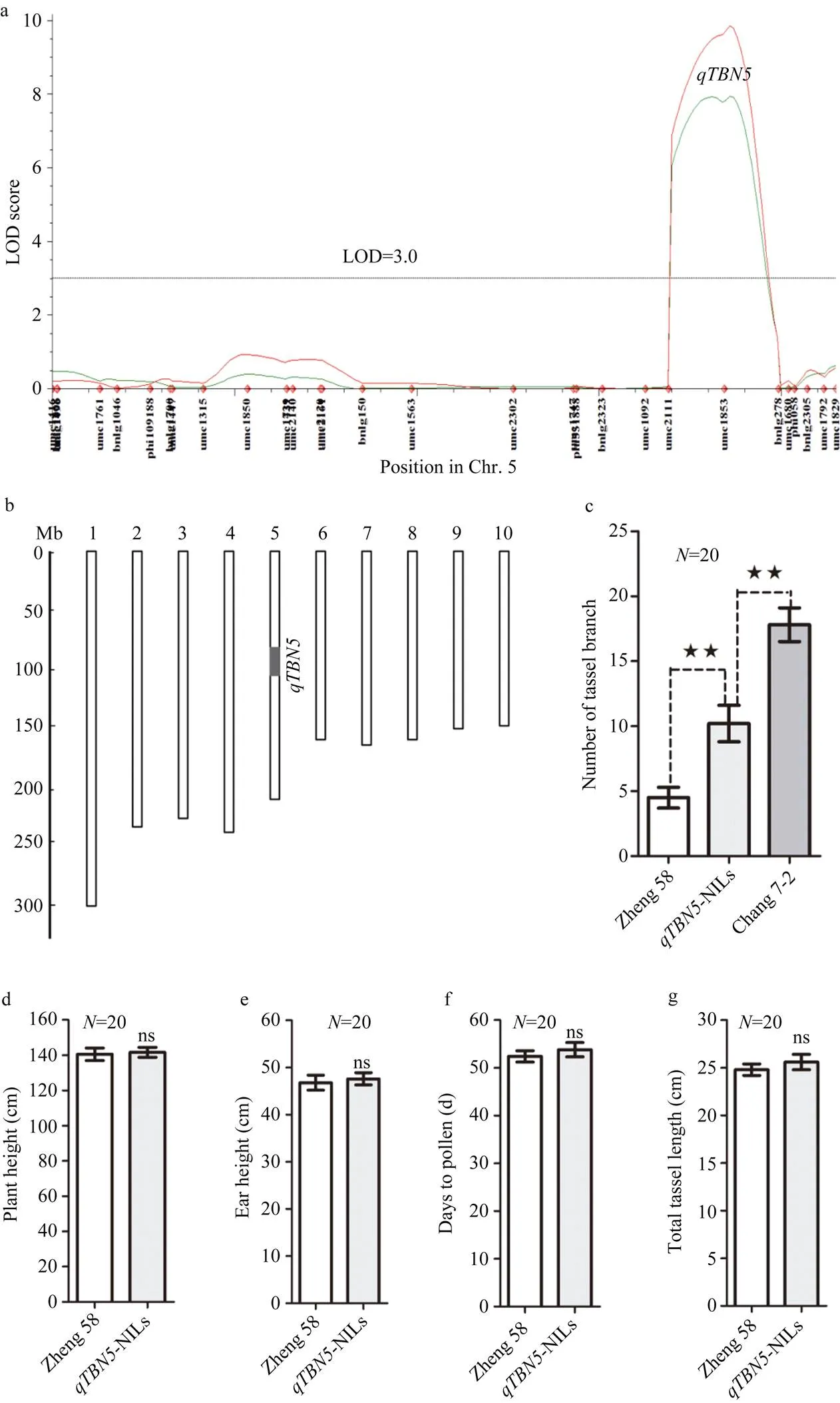
图4 qTBN5-NILs的基因型和表型效应
a: 雄穗分枝数主效基因的定位; b:-NILs的基因型, 白色为郑58背景, 黑色部分为来自昌7-2的基因所在片段; c: 雄穗分枝数; d: 株高; e: 穗位高; f: 抽雄期; g: 雄穗主轴长。**表示在 0.01水平差异显著, ns表示差异不显著。
a: physical locus ofidentified in RILs population. b: genotypes of-NILs. Black part is segment from Chang 7-2 indicating gene position and white parts are genetic background from Zheng 58. c: tassel branch number. d: plant height. e: ear height. f: days to pollen. g: total tassel length.Error bars represent SD. “**” and“ns” show significance at<0.01 level and on significant difference, respectively as determined by-test.
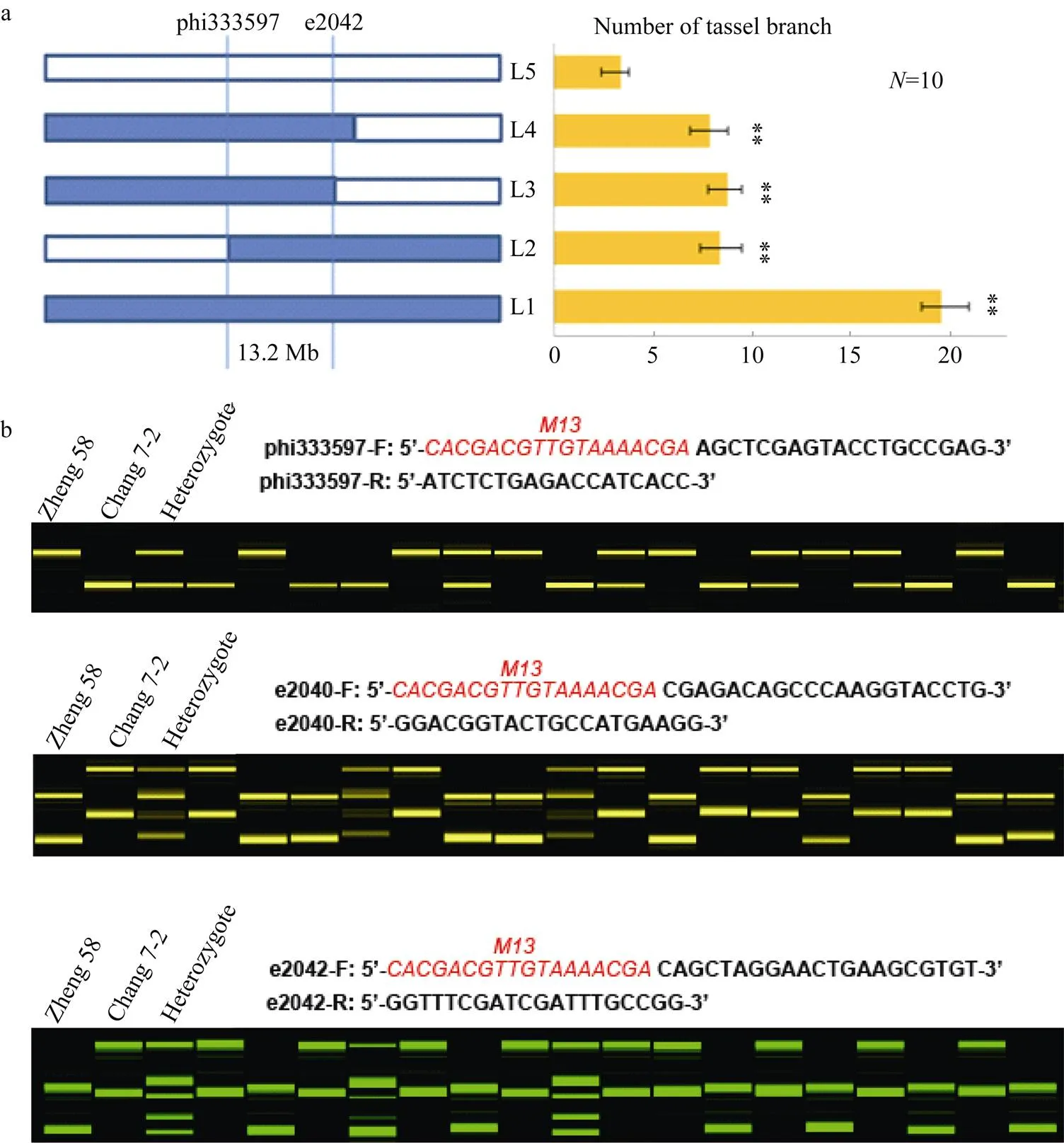
图5 qTBN5进一步定位及连锁标记开发
a: 交换株代换作图定位, L1为昌7-2, L5为郑58, L2~L4为重组株, **表示在 0.01水平差异显著; b: 标记检测郑58和昌7-2及BC1F2世代部分单株基因型, 红色M13为通用引物序列。
a:detected on the substituted segments in recombinant lines; L1: Chang 7-2; L5: Zheng 58. L2–L4: recombinant lines; “**” shows significance at<0.01 as determined by-test. b: genotypes identification ofin BC1F2backcross population. M13: Red indicates universal primer sequences.
3 讨论
玉米雄穗分枝数是复杂的数量性状, 玉米雄穗分枝数QTL定位易受遗传背景、群体类型、标记密度和试验环境等因素影响, 迄今对控制玉米雄穗分枝数的基因研究报道相对较少且存在不同的看法[24]。不同的研究者利用不同研究材料、不同的标记密度, 在玉米10条染色体上都定位出了玉米雄穗分枝数的QTL[4-14]。本研究共检测到5个均可重复检测的主效QTL, 分别位于第3、第4、第5、第7、第10染色体上(图3), 各QTL座位的表型贡献率变异范围在5.81%~19.92%之间(表2)。其中第3染色体定位区间位于[18]和[19]基因附近, 第7染色体定位区间附近有[15]突变体基因, 而定位的区间与Chen等[12]报道区间有少部分重合, 且该区间附近存在[21]突变体基因。对于第5染色体上雄穗分枝数QTL定位的研究, 由于受群体类型及标记密度的影响, 不同的研究中没有相对一致的定位区间, 且效应都比较小[6,9-10,12,14,25], 本研究中定位到的贡献率在5个主效QTL中最大, 达到19.92%。而位于第10染色体的为一个新的QTL, 在此区域未见有QTL报道。因此, 尽管定位到的部分QTL与前人报道的具有重合定位区间, 但由于效应大小及稳定性未知, 仍然需要进行遗传效应验证。
近等基因系和染色体片段代换系是在受体的遗传背景中代换某个或某些供体亲本的染色体片段, 而基因组的其余部分均与受体亲本相同[26], 可对复杂数量性状进行有效的遗传剖析, 因此在基因(QTL)的鉴定与定位, 等位基因变异分析等方面具有重要的利用价值而受到许多研究者的重视。玉米雄穗分枝数是受多基因控制的数量性状, 且存在主效QTL, 然而前人研究中所用到的F2、RIL、BC1S1等群体类型易受遗传背景的影响, 使得彼此定位结果存在差异, 难以通过精细定位克隆雄穗分枝数基因。而本研究根据定位到的控制玉米雄穗分枝数主效QTL, 构建主效基因近等基因系, 有效解决了遗传背景的干扰, 且通过发展近等基因系对该基因遗传效应进行了验证(图4), 开发了紧密连锁标记, 并将进一步定位在13.2 Mb区间之内(图5), 为玉米雄穗分枝数基因精细定位和分子标记辅助育种提供参考。
玉米雄穗分枝数作为衡量雄穗大小的重要指标,是玉米育种与种子生产过程中被研究的与产量形成有关的重要农艺性状之一。育种实践中常常要求母本具有较小雄穗或者较少雄穗分枝数, 这样可以减少对雌穗能量的竞争, 父本一般要求较为发达的雄穗或者较多雄穗分枝数, 这样可以保证充足的粉量以便提高制种产量, 而二者组配的杂交种往往需要具有适度较少雄穗分枝数, 从而达到最优组合[27]。郑单958作为黄淮海地区的主推品种, 其亲本郑58和昌7-2两个优良自交系也一直是研究的热点, 二者之所以在制种过程中获得较高产量的杂交种, 重要原因之一是亲本之间雄穗的合理搭配。本研究通过1928个分子标记筛选, 最终找到288个在郑58和昌7-2之间具有多态性的分子标记, 并对标记的物理位置进行了注释, 建立了一张可以借助ABI 3500XL遗传分析仪高通量检测郑单958品种的指纹图谱, 为后续开展郑58和昌7-2之间包括雄穗分枝数在内的优良性状基因挖掘, 进而解析郑单958品种之所以能够成为主推品种的奥秘奠定基础。
4 结论
定位到5个玉米雄穗分枝数主效QTL, 分别位于玉米5条染色体上, 3个QTL表型贡献率超过了10%。构建了玉米雄穗分枝数主效QTL近等基因系, 验证了基因遗传效应, 并将进一步定位在13.2 Mb区间之内, 为玉米雄穗分枝数主效基因的精细定位和分子育种实践奠定了基础。
[1] Geraldi I O, Miranda Filho J B, Vencovsky R. Estimates of genetic parameters for tassel characters in maize (L.) and breeding perspectives., 1985, 30: 1–14
[2] Upadyayula N, Silva H S, Bohn M O, Rocheford T. Genetic and QTL analysis of maize tassel and inforescence architecture., 2006, 112: 592–606
[3] Gue R, Wasson C. Genetic analysis of tassel size and leaf senescence and their relationship with yield in two tropical low land maize populations., 1996, 4: 275–281
[4] Berke T, Rocheford T. Quantitative trait loci for tassel traits in maize., 1999, 39: 1439–1443
[5] Mickelson S M, Stuber C S, Senior L, Kaeppler S M. Quantitative trait loci controlling leaf and tassel traits in a B73lMo17 population of maize., 2002, 42: 1902–1909
[6] 汤华, 严建兵, 黄益勤, 郑用琏, 李建生. 玉米5个农艺性状的QTL定位. 遗传学报, 2005, 32: 203–209 Tang H, Yan J B, Huang Y Q, Zheng Y L, Li J S. QTL mapping of five agronomic traits in maize., 2005, 32: 203–209 (in Chinese with English abstract)
[7] Upadyayula N, Silva H S, Bohn M O, Rocheford T. Genetic and QTL analysis of maize tassel and inforescence architecture., 2006, 112: 592–606
[8] 高世斌, 赵茂俊, 兰海, 张志明. 玉米雄穗分枝数与主轴长的QTL鉴定. 遗传, 2007, 29: 1013–1017 Gao S B, Zhao M J, Lan H, Zhang Z M. Identification of QTL associated with tassel branch number and total tassel length in maize.(Beijing), 2007, 29: 1013–1017 (in Chinese with English abstract)
[9] 王迪, 李永祥, 王阳, 刘成, 刘志斋, 彭勃, 谭巍巍, 张岩, 孙宝成, 石云素, 宋燕春, 王天宇, 黎裕. 控制玉米雄穗分枝数目和雄穗重的主效QTL的定位. 植物学报, 2001, 46: 11–20 Wang D, Li Y X, Wang Y, Liu C, Liu Z Z, Peng B, Tan W W, Zhang Y, Sun B C, Shi Y S, Song Y C, Wang T Y, Li Y. Major quantitative trait loci analysis of tassel primary branch number and tassel weight in maize (L.)., 46: 11–20 (in Chinese with English abstract)
[10] Brown P J, Upadyayula N, Mahone G S, Tian F, Bradbury P J, Myles S, Holland J B, Flint-Garcia S, McMullen M M, Buckler E S, Rocheford T R. Distinct genetic architectures for male and female inflorescence traits of maize., 2011, 7(11): e1002383
[11] 杨钊钊, 李永祥, 刘成, 刘志斋, 李春辉, 李清超, 彭勃, 张岩, 王迪, 谭巍巍, 孙宝成, 石云素, 宋燕春, 王天宇, 黎裕. 基于多个相关群体的玉米雄穗相关性状QTL分析. 作物学报, 2012, 38: 1435–1442 Yang Z Z, Li Y X, Liu C, Liu Z Z, Li C H, Li Q C, Peng B, Zhang Y, Wang D, Tan W W, Sun B C, Shi Y S, Song Y C, Wang T Y, Li Y. QTL analysis of tassel-related traits in maize (L.) using multiple connected populations., 2012, 38: 1435–1442 (in Chinese with English abstract)
[12] Chen Z L, Wang B B, Dong X M, Liu H, Ren L H, Chen J, Hauck A, Song W B, Lai J S. An ultra-high density bin-map for rapid QTL mapping for tassel and ear architecture in a large F2maize population., 2014, 15: 433
[13] Wu X, Li Y X, Shi Y S, Song Y C, Zhang D F, Li C H, Buckler E S, Li Y, Zhang Z W, Wang T Y. Joint-linkage mapping and GWAS reveal extensive genetic loci that regulate male inflorescence size in maize., 2016, 14: 1551–1562
[14] Xu G H, Wang X F, Huang C, Xu D Y, Li D, Tian J G, Chen Q Y, Wang C L, Liang Y M, Wu Y Y, Yang X H, Tian F. Complex genetic architecture underlies maize tassel domestication., 2017, 214: 852–864
[15] Vollbrecht E, Springer P S, Goh L, Buckler E S, Martienssen R. Architecture of floral branch systems in maize and related grasses., 2005, 436: 1119–1126
[16] Satoh-Nagasawa N, Nagasawa N, Malcomber S, Sakai H, Jackson D. A trehalose metabolic enzyme controls inflorescence architecture in maize., 2006, 441: 227–230
[17] Bortiri E, Chuck G, Vollbrecht E, Rocheford T, Martienssen R, Hake S.encodes a LATERAL ORGAN BOUNDARY domain protein that determines the fate of stem cells in branch meristems of maize., 2006, 18: 574–585
[18] Gallavotti A, Zhao Q, Kyozuka J, Meeley R B, Ritter M K, Doebley J F, Pè M E, Schmidt R J. The role ofin the architecture of maize., 2004, 432: 630–635
[19] Walsh J, Freeling M. Thegene of maize functions during the transition from the vegetative to the reproductive shoot apex., 1999, 19: 489–495
[20] Skirpan A, Culler A H, Gallavotti A, Jackson D, Cohen J D, McSteen P. BARREN INFLORESCENCE2 interaction with ZmPIN1a suggests a role in auxin transport during maize inflorescence development., 2009, 50: 652–657
[21] Chuck, G, Brown, P, Meeley, R, Hake, S. Maizetranscription factorsandaffect yield traits by regulating the rate of lateral primordia initiation., 2014, 111: 18775–18780
[22] Lander E S, Green P, Abranhanson J, Barlow A, Daley M, Lincoln S, Newburg L. MAPMAKER: an interactive computer package for constructing primary genetic linkage maps of experimental and natural populations., 1987, 1: 174–181
[23] Meng L, Li H H, Zhang L Y, Wang J K. QTL IciMapping: Integrated software for genetic linkage map construction and quantitative trait locus mapping in bi-parental populations., 2015, 3: 169–173
[24] Brewbaker J L. Diversity and genetics of tassel branch numbers in maize., 2015, 55: 65–78
[25] Briggs W H, McMullen M D, Gaut B S, Doebley J. Linkage mapping of domestication loci in a large maize-teosinte backcross resource., 2007, 177: 1915–1928
[26] Kubo T, Aida Y, Nakamura K, Tsunematsu H, Doi K, Yoshimura A. Reciprocal chromosome segment substitution series derived fromandcross of rice (L.)., 2002, 52: 319–325
[27] Chen Z J, Yang C, Tang D G, Zhang L, Zhang L, Qu J T, Liu J. Dissection of the genetic architecture for tassel branch number by QTL analysis in two related populations in maize., 2017, 16: 1432–1442
Major Quantitative Trait Loci Mapping for Tassel Branch Number and Construction ofNear-isogenic Lines in Maize (L.)
DAI Zi-Ju1, WANG Xin-Tao1, YANG Qing1, WANG Yan1, ZHANG Ying-Ying1, XI Zhang-Ying2, and LI Bao-Quan1,*
1Crop Designing Centre, Henan Academy of Agricultural Sciences, Zhengzhou 450002, Henan, China;2College of Agronomy, Henan Agricultural University / Collaborative Innovation Center ofHenanFood Crops, Zhengzhou 450002, Henan, China
Tassel branch number (TBN) is a principal component of maize tassel-related traits important for modern maize breeding and production. To understand the genetic basis of tassel branch number, we adopted 288 markers exhibiting polymorphisms between Zheng 58 and Chang 7-2 to construct the genetic linkage map, and conduct quantitative trait loci (QTLs) analysis using sets of 188 recombinant inbred line (RIL) families derived from the elite maize inbred lines. Zheng 58 × Chang 7-2. Five major QTLs controlling tassel branch number within five different chromosomes respectively were validated by using the inclusive composite interval mapping method (ICIM) based on phenotypic data collected in two years. Becauselocated on chromosome5.05 was found to be an important QTL,near-isogenic lines (-NILs) were developed by backcrossing and marker-assisted selection, andwas further mapped in 13.2 Mb intervals.These findings will advance our understanding of the inheritance basis of TBN, and also facilitate the fine mapping and molecular breeding programs in maize.
maize; tassel branch number; quantitative trait loci mapping; near-isogenic lines
2018-06-09;
2018-06-11.
10.3724/SP.J.1006.2018.01127
李保全, E-mail: lbq308@126.com, Tel: 0371-65721718
E-mail: zijudai@163.com, Tel: 0371-65867618
2017-12-20;
本研究由河南省重大科技专项(161100110500)和河南省农业科学院科研发展专项(YNK20177514, YNK201710605)资助。
This study was supported by the Henan Major Science and Technology Project (161100110500) and the Special Fund for Scientific Research and Development of Henan Academy of Agricultural Sciences (YNK20177514, YNK201710605).
URL: http://kns.cnki.net/kcms/detail/11.1809.S.20180611.0555.004.html
